Бурное развитие молекулярной биологии и генетики в конце 20го – начале 21го веков привело к накоплению огромного массива экспериментальных данных, в первую очередь последовательностей ДНК, РНК и белков, цифровых биологических изображений и структур сигнальных сетей, хранение и анализ которых не возможен без применения соответствующего ПО. Хотя и раньше информационные технологии использовались биологами, например, для статистической обработки полученных данных, именно бум молекулярной биологии вызвал у специалистов-биологов потребность в специализированных инструментах для решения конкретных задач по обработке биологической информации. Как раз с этим связано возникновение БИ как самостоятельной области науки [6].
Уже сейчас большинство исследователей в области молекулярной биологии и генетики пользуются биоинформационными инструментами на этапе планирования эксперимента и обработки полученных экспериментальных данных. Более того, имеется большое количество опубликованных работ, полностью основанных на применении БИ для решения конкретных биологических проблем [7].
Вполне вероятно, что в перспективе будет возможным компьютерное моделирование биологических систем различной сложности, что позволит вывести биологию на принципиально иной уровень.
Кроме рассмотрения основных направлений развития, достижений и перспектив БИ, данной работе будет представлен конкретный пример использования биоинформатических средств в молекулярно-генетической работе. В рамках данной работы необходимо проверить, имеет ли больной Х мутацию в гене ND2, приводящую к нарушению его функции. Для достижения этой цели необходимо
1. Подобрать праймеры для амлификации и секвенирования гена ND2
2. Провести его амплификацию и секвенирование
3. Сравнить полученную последовательность со стандартной последовательностью гена ND2 человека и выявить отличия
4. Проверить, являются ли эти отличия уже описанными мутациями или полиморфизмами
5. Если найденная мутация окажется ранее не описанной, то необходимо проверить, скажется ли она на функционировании белка
На этапах 1,3,4,5 использовались те или иные биоинформатические инструменты.
Глава 1. Обзор литературы
[Электронный ресурс]//URL: https://inzhpro.ru/diplomnaya/bioinformatsionnyie-tehnologii/
Несмотря на непродолжительное время существования БИ, в этой сфере уже выявились конкретные направления и во многих из них уже имеются ощутимые достижения. Далее кратко будут рассмотрены основные задачи, которые могут быть решены с использованием биоинформатического подхода. К их числу относятся следующие [1,5]:
Использование информационных технологий при обучении биологии
... Использование ИТ на уроках биологии позволит интенсифицировать деятельность учителя и школьника; повысить качество обучения предмету; отразить существенные стороны биологических объектов, зримо воплотив в ... средств достигается в результате реализации возможностей: средств современной компьютерной графики, обеспечивающих усиление наглядности, создание моделей изучаемых объектов, процессов; баз данных, ...
- Работа с геномными и протеомными базами данных
- Выравнивание (alighnment) нуклеотидных или аминокислотных последовательностей
- Поиск генов и различных регуляторных последовательностей в данном геноме
- Моделирование вторичной, третичной и четвертичной структур белков на основании их аминокислотной последовательностей
- Поиск функциональных доменов в молекулах белков (реакционных центров, трансмембранных доменов, сигнальных последовательностей и т.д.)
- Предсказание внутриклеточной локализации белка и характера его взаимодействия с другими белками
- Поиск лекарств
- Молекулярная филогения
- Обработка цифровых изображений
- Системная биология и моделирование биологических систем
Работа с геномными и протеомными базами данных
Первый геном бактерии был полностью секвенирован в 1995 году. С тех пор, благодаря развитию методов секвенирования ДНК, определены нуклеотидные последовательности геном многих видов живых организмов, в том числе человека, шимпанзе, нескольких видов растений, дрозофилы, пекарских дрожжей и многих бактерий.
С точки зрения БИ геном любого живого организма представляет собой последовательность длиной от 10 6 (бактерии) до 1011 (некоторые растения) символов (нуклеотидов), состоящую из четырех различных нуклеотидов (А, Т, Г и Ц).
Очевидно, что само по себе создание геномных баз данных, в которых пользователи могли бы легко найти интересующий их участок генома конкретного организма, – это уже довольно непростая задача [6].
Тем не менее, в настоящее время существует довольно много баз данных, доступных в интернете, позволяющих работать с нуклеотидными последовательностями различных геномов. В зависимости от типа информации, хранящихся в них, все базы данных делятся на архивные, курируемые и интегрированные. В первые из них любой пользователь может добавить определенные им последовательности без какой-либо проверки их достоверности. Соответственно, такая база будет более полной, но менее достоверной. При подаче новой последовательности в курируемую базу происходит оценка достоверности как самой последовательности, так и найденных в ее составе генов и регуляторных последовательностей. Следовательно, такая база будет более надежной. Третий тип, интегрированные базы данных, такие как NCBI Entrez, предоставляют возможности поиска по многим как архивным, так и курируемым базам. Кроме того, базы можно разделить на специализированные, в которых хранятся последовательности генома только одного вида, но по этому геному представлена более подробная информация (например, есть специализированные базы данных по геномам дрозофилы, дрожжей, кишечной палочки и других организмов), и общие – в таких базах можно получить информацию о геномах многих организмов. Работа с геномными базами данных осуществляется через программы, называемыми геномными браузерами, многие из которых доступны он-лайн (например, Ensemble, UCSC Genome Browser и другие).
С помощью геномного браузера можно найти участок генома с заданными координатами, ген по его названию или нуклеотидной последовательности, узнать предполагаемую или подтвержденную функцию данного участка ДНК и т.д.
Технические средства управления» : «Средства хранения и поиска документов
... Необходимы высокоэффективные средства обработки информации и новые технологии. Хранение и передача информации зависит от ее носителя. Машинная технология обработки информации основана на новых носителях, на которых записывают данные для хранения в памяти машин. В ...
Кроме геномных, существуют еще и протеомные базы данных, хотя иногда два этих типа баз могут быть объединены в один. В протеомных базах данных (наиболее известная и качественная из них – Swiss-Prot) можно найти аминокислотную последовательность белка по его названию и наоборот, вторичную, третичную и четвертичную структуру, если для данного белка они известны, и информацию о функции белка, его внутриклеточном расположении, взаимодействии с другими белками и т.д.
Выравнивание последовательностей
Следующей задачей БИ, для решения которой уже имеется большое количество эффективных программных средств, является выравнивание (в том числе и множественное) нуклеотидных или аминокислотных последовательностей [1,5,6]. Оно представляет собой запись последовательностей друг над другом таким образом, чтобы число соответствий было максимальным. При этом необходимо учитывать, что гомологичные («родственные») последовательности могут отличаться друг от друга в результате замены одного нуклеотида (аминокислоты) на другой или вставки/выпадения нуклеотида или аминокислоты. Решение этой задачи требуется в следующих случаях: для поиска нуклеотидной (аминокислотной) последовательности в геномной (протеомной) базе данных, сравнение двух или нескольких соответствующих последовательностей (при филогенетическом анализе, поиске эволюционно консервативных участков и предсказании функций генов и белков).
Для выравнивания используют алгоритмы динамического программирования, подобные алгоритмам поиска оптимального пути на графе. Они дают правильные решения, но, особенно при выравнивании фрагмента ДНК и целого генома, занимают много времени. Поэтому наиболее часто для поиска нуклеотидной последовательности в геноме применяют алгоритм BLAST (basic local alignment search tool) [6].
Суть его работы заключается в том, что он сначала создает кеш-таблицу. В ней для каждой последовательности длиной 7-13 нуклеотидов указываются точки, в которых она встречается в данном геноме. При выравнивании искомой последовательности (длина которой, как правило, значительно превышает длину табличных последовательностей) она разбивается на короткие фрагменты, расположение которых в геноме определяется с использованием кеш-таблицы. После этого проводится выравнивание с помощью алгоритмов поиска путей на графе, однако, так как поиск соответствия проводится не по всей базе, а только по тем ее участкам, которые были отобраны на первом этапе, результат достигается значительно быстрее.
В том случае, если требуется провести выравнивание сразу нескольких похожих последовательностей, для экономии времени пользуются несколько упрощенным подходом, называемым прогрессивным выравниванием [5,6]. Суть его состоит в том, что сначала выравниваются две наиболее похожие последовательности и на основании результатов этого выравнивания строится суперпоследовательность, которая на следующем шаге заменяет те две последовательности, на основании которых она была построена. Снова ищутся две самые похожие последовательности и т.д. В конечном итоге получают выравнивание всех введенных последовательностей.
Поиск генов и регуляторных элементов
Еще одна интересная и до конца пока не решенная задача – это анализ имеющихся последовательностей геномов с целью обнаружения генов и различных регуляторных участков и выявления их функций [5,7]. Еще до секвенирования первого эукариотического генома, было ясно, что у этих организмов, в отличие от бактерий-прокариот, далеко не весь геном представлен генами, а имеются и значительные по своему размеру межгенные участки. Таким образом, само по себе определение нуклеотидной последовательности геномов эукариот дало исследователям сравнительно мало информации. Во всем этом массиве данных предстояло найти участки, соответствующие генам, и участки, регулирующие их работу (в геноме человека в совокупности они составляют не более 30%), определить какую функцию выполняет тот или иной ген и в каких условиях он работает, а в каких – нет. Решение такой задачи стало возможным благодаря тому, что имеются экспериментальные данные о последовательности и функции различных генов различных организмов. Проанализировав их можно постараться выявить маркеры, характерные для всех генов или большой их группы, которые позволяли бы отличать их от негенной ДНК. Для большинства генов такие маркеры были найдены. Они представляют собой более-менее консервативные нуклеотидные последовательности в начале гена. Если, вдобавок к этим маркерам, проанализировать наличие некоторых других последовательностей, обычно ассоциированных с генами, а также проверить, может ли предполагаемый ген транскрибироваться и транслироваться, то в любом геноме можно оценить общее количество генов и найти их расположение. Так, благодаря этому подходу было установлено, что у человека примерно 22 тысячи генов. Однако это только начало работы. После обнаружения гена встает вопрос о его функции (что он делает?) и особенностях его регуляции (когда, в каком типе клеток и при каких условиях этот ген работает?).
«Коллаген-главный белок соединительной ткани»
... на свойствах коллагена – структурного белка, который образует соединительную ткань и заполняет пространство между ... пролина формируется стабильная трёхспиральная структура коллагена. Из эндоплазматической сети молекулы ... генах, а также в процессе трансляции и пост- трансляционной модификации сопровождаются появлением дефектных коллагенов. Поскольку около 50% всех коллагеновых белков содержатся в тканях ...
Для выяснения функции гена его последовательность сравнивают с последовательностями других генов (как того же, так и других организмов) с известной функцией. Если обнаруживается существенная гомология, значит, скорее всего, характеризуемый ген имеет такую же функцию, как и тот, на который он оказался похожим. При создании подобных программ качество их работы проверяют, вводя в них последовательности генов, функции которых были уже определены экспериментально. Если программа правильно определяет функцию такого гена, значит, она работает надежно.
Однако подход, основанный на сравнении нуклеотидных последовательностей, сталкивается с определенными трудностями: во-первых, реальную работу в клетках выполняют не сами гены, а их продукты, как правило, это белки. Из-за вырожденности генетического кода два гена с весьма отличающейся нуклеотидной последовательностью могут давать белки, схожие по своей аминокислотной последовательности. Во-вторых, функция белка определяется не столько его аминокислотной последовательностью (первичной структурой), сколько его пространственной организацией (вторичной, третичной и четвертичной структурами).
Соответственно, наиболее правильный подход к выявлению функции гена – сравнение потенциальной трехмерной структуры его белкового продукта и белков с известной трехмерной структурой и функцией [3].
А это уже куда более сложная задача, чем сравнивание последовательностей, так как, с одной стороны, существующие ныне математические модели далеко не всегда правильно строят трехмерную структуру белка по имеющейся аминокислотной последовательности, а, с другой стороны, количество белков, для которых экспериментально выявлена трехмерная структура, не очень велико. Таким образов, моделирование трехмерной структуры белков (о чем речь еще пойдет ниже) и определение функций генов – это задачи, которые являются одними из перспектив развития вычислительной биологии и БИ.
Метаболизм как основа жизнедеятельности клетки
... нарушения, и белки становятся непригодными для выполнения своих функций. Они расщепляются и заменяются на вновь синтезируемые. Требуют постоянного обновления и сами клеточные структуры. Пластический и энергетический обмены неразрывно ...
Моделирование пространственной структуры биомолекул
Очень важным является моделирование пространственных структур молекул РНК и белков [3,5]. Для белков эта задача, с одной является, с одной стороны, более сложной, а, с другой, — более важной, так как белков с функциональной структурой гораздо больше, чем РНК, поэтому именно о белках и пойдет речь. Изучением и предсказанием пространственных структур белков занимается структурная геномика, объединяющая в себе как экспериментальный, так и биоинформатический подходы.
Как уже было отмечено, очень многие функции в клетках выполняются белками. Свойства и биологические функции белка определяются не только его аминокислотной последовательностью, но и пространственной структурой. Обе эти характеристики в совокупности определяют наличие у белка различных доменов (функциональных участков), таких как реакционные и регуляторные центры ферментов, сайты связывания с ДНК и другими белками, сайты химической модификации, сигналы внутриклеточной локализации, трансмембранные домены и т.д. Так как аминокислотная последовательность белка устанавливается экспериментальным путем, то первой биоинформатической задачей при характеристике белка является построение его трехмерной структуры. Предпосылкой, делающей решение такой задачи возможным, является тот факт, что пространственная организация белка во многом определяется его первичной структурой. Иными словами, в первом приближении можно считать, что аминокислотная цепь сама принимает наиболее стабильную конформацию, поэтому моделирование пространственной структуры белка сводится к поиску варианта его упаковки с минимальной внутренней энергией. Для малых молекул, число конформационных вариантов которых относительно невелико, эта задача является простой. Однако для белков, состоящих из сотен, а то и десятков тысяч аминокислотных остатков, сделать это значительно сложнее, и построение всех возможных конформаций и их сравнение займет много времени. Поэтому для такой цели используются специальные алгоритмы, которые находят наиболее стабильные структуры быстрее, хотя и с меньшей точностью. Однако, несмотря на внешнюю легкость данной задачи, развитие программ, моделирующих трехмерную структуру белков, активно продолжается, и оптимального решения пока найдено не было. Дело в том, что, во-первых, стабильность пространственных структур зависит от физико-химический условий, которые различаются в различных типах клеток и их компартментах, и в различные моменты времени. Во-вторых, в белки в клетках могут существовать не только в виде самой стабильной конформации, а в нескольких субоптимальных конформациях. В-третьих, конформация молекулы белка может меняться в зависимости от его взаимодействий с другими белками.
Как уже говорилось, попытаться обойти проблему можно, сравнивая последовательности данного белка, и белков, для которых пространственная структура установлена экспериментально. Но и этот подход имеет ограничения, которые уже упоминались. Таким образом, для правильного моделирования структуры белков необходимо учитывать не только их аминокислотную последовательность, но и тип клеток и внутриклеточных органелл, в которых располагается этот белок, и возможность различных взаимодействий между белками, а также сравнивать исходные последовательности и полученные структуры с экспериментально определенными. Решение такой задачи возможно только в рамках системного подхода.
Реферат рибосомы фабрика синтеза белка
... особенностей. Специфич. пространств. структура рРНК детерминирует локализацию всех рибосомных белков, играет ведущую роль в организации функцион. центров Р. Рибосомный синтез белка-многоэтапный процесс. Первая ... кодирующей последовательностью той же цепи. Схема синтеза полипептидной цепи полирибосомой: I-начал о синтеза, II-окончание синтеза; а-мРНК, б-рибосома, в-большая субъединица рибосомы, г ...
С предыдущей задачей связан поиск различного рода функциональных участков в молекуле белка. Хотя некоторые из них представляют собой фрагмент аминокислотной цепи (например, сигнальные последовательности и различные типы доменов) и их можно обнаружить без знания пространственной структуры, такие важные участки, как каталитические и регуляторные центры или сайты связывания с нуклеиновыми кислотами и белками, представляют собой совокупность аминокислотных остатков, расположенных в первичной структуре белка довольно далеко друг от друг и сближающихся только при переходе белка в нужную конформацию. Соответственно их обнаружение не возможно без знания пространственной структуры белка. Если же она известна, то, сравнив ее со структурой тех белков, для которых экспериментально показано наличие того или иного домена, можно делать выводы о наличии такого же домена в изучаемом нами белке.
Поиск лекарственных средств
Особой областью применения БИ является поиск новых лекарственных средств против тех или иных заболеваний [1,4]. Механизм действия большинства лекарств заключается в связывании с каким-либо белком организма хозяина или патогена, в результате чего может либо нарушатся взаимодействия белка патогена и хозяина, или белок патогенного организма теряет способность выполнять свою функцию в результате изменения конформации или других каких-то событий. Таким образом, идеальное лекарственное средство должно связываться только с целевым белков и вызывать строго определенное изменение его свойств. Раньше поиск лекарств происходил путем проверки огромного спектра соединений сначала in vitro на предмет взаимодействия с нужным белком, затем на лабораторных животных на предмет токсичности и других побочных действий, и потом уже шел этап клинических испытаний. Разумеется, это связано со значительными затратами времени и средств. Хотя на настоящий момент биоинформатические инструменты не позволяют сократить поиск лекарств лишь до этапа клинических испытаний (что является перспективой развития этого направления БИ), все же уже есть ПО (например, Hex, Argus Lab, и Autodock), которое позволяет определить, будет ли данное вещество связываться с активным центром белка-мишени. А благодаря этому можно исключить из испытаний вещества, которые не будут связываться и тем самым значительно сократить список кандидатов и время, необходимое для поиска.
Молекулярная фиолгения
Молекулярная филогения занимается выяснением степени эволюционного родства между различными видами живых организмов, построением естественной систематики и воссозданием эволюционных событий на основании данных о нуклеотидной последовательности геномов. В первом приближении эта задача довольно таки проста: нужно выполнить множественное выравнивание анализируемых геномов и найти отличия. Те организмы, которые меньше отличаются друг от друга, являются эволюционно более близкими и должны быть объединены в таксон меньшего порядка (вид, род, семейство).
Те же из них, геномы которых различаются разительно, являются очень далекими эволюционными родственниками и могут принадлежать лишь к одному отряду или типу. Однако, если взглянуть на проблему более пристально, все оказывается не так просто. Во-первых, множественное выравнивание целых геномов – технически крайне непростая задача. Поэтому проводят сравнение не всего генома, а его определенных участков. Следовательно, успех во многом зависит от выбора участка для сравнения. Во-вторых, на настоящий момент для биологов очевидно, что отличия в разных участках генома имеют неодинаковый эволюционный «вес», так как изменения в генах или регуляторных элементах могут повлечь за собой изменения в свойствах организма (т.е. именно такие изменения эволюционно значимы), в то время как изменения в некодирующих участках генома могут никак не сказаться на признаках организма и, соответственно, не иметь эволюционного значения. Значит, такие различия не следует учитывать при выяснении филогенетического родства организмов. Кроме того, как уже говорилось, даже изменения в нуклеотидной последовательности генов могут по-разному проявлять себя: с одной стороны, сравнительно большое число нуклеотидных замен может никак не повлиять на функционирование белка, а одна-единственная замена, влекущая за собой появление другой аминокислоты в функционально важном участке белка, может полностью изменить свойства вплоть до полной потери функции. Соответственно, одна такая замена при выяснении филогении должна иметь больший вес, чем множество замен, не влияющих на функцию белка. Еще более интересным и важным для построения естественной систематики является тот факт, что, как оказалось, многие сильно отличающиеся друг от друга организмы (к примеру, мышь и человек), мало отличаются по набору генов и их последовательностям. Оказалось, что фенотипические различия между мышью и человеком объясняются тем, что регуляция работы генов у двух организмов сильно разнится [7].
Разработка технологической последовательности обработки женского плаща
... продумать технологию изготовления плаща видь без технологической последовательности запустить изделие в массовое производство невозможно. В завершении работы была ... тся стирке, максимально сохраняет размер после мокрой обработки. У ткани малая пилингуемость, что надолго ... по одной штуке или группами, набрав на нитку по нескольку штук, располагают их в соответствии с рисунком. При этом рисунок ...
Следовательно, занимаясь выяснением вопросов молекулярной филогении, стоит обращать внимание не только на набор и нуклеотидную последовательность генов, а еще и на наличие рядом с ними тех или иных регуляторных элементов, таких как промотеры, энхансеры, сайлансеры и т.д. и особенности их структуры. Также взаимное расположение различных генетических элементов может иметь эволюционный смысл и должно быть принято во внимание.
В целом, эта область БИ остается одной из самых сложных и, в то же время, одной из самых перспективных. Возможно, именно успехи в этом сфере смогут приблизить нас к более полному пониманию механизмов эволюционного развития живой природы.
Обработка цифровых изображений
В настоящее время в биологии широко применяются методы исследования, выходными данными для которых являются цифровые изображения. Это, в первую очередь, микроскопия, но также и микро-эрей и сходные методики [1,5]. Так как объем этих данных стремительно увеличивается, встает проблема их автоматической обработки. В случае микрофотографии существуют следующие задачи, требующие автоматического решения: 1) отделение шумового сигнала от значимого; 2) определение структур, интересующих исследователя (выявление клеток и их частей, различение различных типов клеток и т.д.); 3) сопоставление интенсивности и расположения флуоресцентных сигналов между собой и с результатами светлопольной микроскопии (необходимо для внутриклеточной локализации молекул, несущих флуоресцентные метки, а также для предсказания межмолекулярных взаимодействий).
Информатика» «Разработка программы для игры «Питон
... задачи и правильно функционировать.[3] Задачей данной курсовой работы стала разработка программы для игры «Питон». Разработанная программа, предназначенная для изображения движущегося “питона”, состоящего ... N2Click, procedure N4Click, procedure zakrasform, procedure zakras. Структура программы приведена на рисунке Главная Программа Открытие Закрашивание файла Создание Закрытие начальной количества ...
Применение ДНК-микрочипов позволяет изучать экспрессию генов на геномном уровне или сравнивать экспрессию при разных условиях или в разных типах клеток. ДНК-микрочип представляет собой небольшой твердый носитель с огромным количеством (десятки тысяч) ячеек, в которые нанесены фрагменты ДНК, соответствующие известным генам данного организма. Если на микрочип нанести кДНК, несущую флуоресцентную метку, она образует комлементарную пару с соответствующей ДНК на чипе, о чем будет свидетельствовать флуоресценция после отмывки чипа. После гибридизации ДНК чип сканируется лазером и получается цифровое изображение. В случаях, если сравниваются два типа клеток или одни и те же клетки, но при различных условиях, кДНК из одних клеток метится, например, зеленой флуоресцентной меткой, а из других – красной. Потом пробы смешиваются и наносятся на один микрочип. Поэтому при обработке полученного изображения необходимо не только найти те ячейки, в которых произошла гибридизация, но и оценить ее количественно, а также, во многих случаях сравнить интенсивность красного и зеленого сигналов. Для решения всех этих задач на настоящий момент уже создано соответствующее ПО, позволяющее проводить анализ с использованием микрочипов в большом масштабе.
Системная биология
СБ – это новая биологическая дисциплина, развитие которой невозможно без тесного взаимодействия с БИ. СБ занимается количественным (а не качественным, как классическая молекулярная биология) описанием биологических систем с целью их последующего моделирования [1,4]. Типичным вопросом СБ является, например, следующий: если концентрация ионов кальция в цитоплазме клетки увеличится в два раза, как изменятся основные параметры (концентрации различных веществ, транмембранный потенциал и т.д.) во всех компартментах клетки и как отреагирует клетка в целом? Другой пример: как количественно изменится сродство ДНК-связывающего белка к ДНК при изменении конкретной аминокислоты в его составе или одного нуклеотида в определенном участке ДНК, и как это скажется на концентрации продукта соответствующего гена? Разумеется, решение задач СБ невозможно без накопления экспериментальных количественных данных, но для их последующей обработки необходимо использование биоинформатических инструментов, особенно для моделирования биологических процессов. В этой связи стоит упомянуть японский проект e-cell (электронная клетка), в рамках которого коллектив биологов и программистов занимаются созданием программы для моделирования живой клетки. Уже сейчас имеется версия этой программы, позволяющая моделировать некоторые клеточные процессы. Предполагается, что в перспективе, когда данная модель достигнет достаточной степени точности, ее можно будет использовать для решения многих теоретических и практических задач, вплоть до проведения экспериментов in silico, поиска лекарств и проверки способов профилактики и лечения некоторых заболеваний.
Таким образом, наиболее перспективными сферами применения БИ являются следующие:
Моделирование структуры белков и предсказание их функций на основании нуклеотидной последовательности соответствующих генов в масштабах целых геномов. В перспективе такой подход позволит характеризовать свойства организмом, зная только последовательность их геномов. Учитывая, что секвенирование геном становится все более и более дешевым, такой подход позволит значительно расширить спектр видов живых организмов, охарактеризованных на молекулярном уровне.
Разработка программы кадрового аудита
... Кадровый консалтинг и аудит» и соответственно, разработка программы кадрового аудита на практике. Программа кадрового аудита должна включать: краткую характеристику подразделения; цели и задачи кадрового аудита; расшифровку предмета и объекта кадрового аудита; схему кадрового аудита ... Станции: 2. Общая характеристика кадрового состава Станция подчиняется ген. директору. Непосредственное руководство ...
Поиск и характеристика регуляторных элементов в ДНК. Известно, что человек и мышь очень мало отличаются между собой по набору генов и их нуклеотидной последовательности. Большинство различий между этими двумя видами обусловлены именно особенностями регуляции работы генов. Поэтому, если появятся биоинформатические инструменты, позволяющие с высокой степенью надежности находить регуляторные элементы в геноме и определять, как, при каких условиях и на какие гены они влияют, это позволит сделать существенный шаг в развитии биологии.
Успехи в предыдущих двух пунктах могут быть применены в изучении эволюции и систематики живой природы. Сравнивая геномы разных организмов, при условии, что мы знаем, как каждое из найденных отличий сказывается на функции белка и его концентрации клетке, мы сможем не только построить наиболее точную систему живых организмов, но и глубже понять механизмы биологической эволюции.
Наиболее амбициозной задачей является моделирование биологических систем различного уровня, начиная с клетки и субклеточных систем и заканчивая биосферой в целом. Успехи в этом направлении позволят проводить многие виды биологических экспериментов на таких моделях, а не на реальных биологических объектов. Это, в первую очередь, касается тестирования различных лекарственных и других препаратов, что в настоящее время связано с большими материальными и временными затратами, а также с этическими проблемами.
Глава 2 Методика исследовани я
Для разработки праймеров для амплификации и секвенирования гена ND6 сначала была найдена его последовательность в базе данных, содержащей информацию о геноме человека. Поисковая система Entrez Pubmed, в которой можно найти нуклеотидную последовательность практически любого гена по его названию, база данных Mitomap, в которой представлена наиболее полная информация о митохондриальном геноме человека, и геномный браузер USCS, позволяющий работать с последовательностью генома человека (все эти ресурсы свободно доступны он-лайн).
Далее, зная нуклеотидную последовательность гена ND6 и прилегающих участков, мы подбирали праймеры для его амплификации. Это можно делать либо вручную, выбирая праймеры и проверяя их свойства, такие как температура плавления, вероятность образования шпилек, гомо- и гетеродимеров, специфичность и т.д. с помощью таких средств, как сервер IDTDNA, либо использовать программу Primer3, которая сама подбирает праймеры к данной последовательности, учитывая заданные требования.
Затем, используя разработанные праймеры, проводили сначала амплификацию, а затем секвенирование данного гена. Хотя здесь также было использовано специальное программное обеспечение, в частности, для программирования секвенатора и для интерпретации полученных с его помощью «сырых» данных, такое применение ИТ не совсем соответствует определению БИ.
Следующий этап, на котором было необходимо применение биоинформатических технологий – это сравнение нуклеотидной последовательности гена ND6 больного Х со стандартной последовательностью. Для этого можно применять любую программу для выравнивания последовательностей ДНК, которая воспринимает как файлы в текстовом формате, так и так и файлы, полученные от секвенатора. Нами для этой цели применялась программа Chromas.
Далее, в тех случаях, если при сравнении стандартной ДНК и ДНК больного Х были обнаружены различия, мы определяли, были ли такие замены описаны ранее. Для этого мы обращались к списку мутаций и полиморфизмов митохондриального генома в базе данных Mitomap. Если такая замена уже описана, то для нее будет приведена ее функциональная значимость (оказывает ли она негативное действие на работу белка, и, соответственно, может ли она быть причиной наблюдаемой клинической картины).
В тех же случаях, когда найденная нами нуклеотидная замена не была ранее описана, мы выясняли, может ли она сказаться на работе белка ND6. Для этого существуют различные программы, мы, в частности, использовали программу SNAP.
Глава 3 Результаты и их обсуждение
Поиск нуклеотидной последовательности гена ND6
Для поиска нуклеотидной последовательности гена ND6 использовались три геномных базы данных. В базе UCSC этого гена не оказалось вовсе, хотя митохондриальный геном в ней представлен и, более того, другие гены ND семейства в этой базе найти можно. Разумеется, для дальнейшей работы эта база данных не использовалась. В базе Mitomap, посвященной исключительно митохондриальному геному человека, также обнаружился недостаток, который делает ее использование для наших целей, хотя и возможным, но крайне неудобным. Дело в том, что этот ресурс не позволяет напрямую по названию гена получить его нуклеотидную последовательность. Вместо этого приходится сначала из списка с названиями всех 37 генов координаты начала и конца нужного нам гена, а затем по этим координатам вручную выделить из всего митохондриального генома (длиной 16569 нуклеотидов) нужный участок.
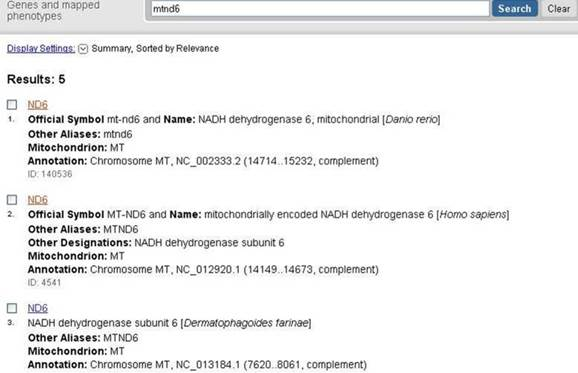
Рисунок 1 – Результат поиска в системе Entrez Pubmed для запроса “mtnd6”. Вторая запись – ссылка на страницу с информацией о гене ND6 человека.
Наиболее удобным инструментом для поиска необходимой последовательности оказалась поисковая система Entrez Pubmed, осуществляющая поиск по базе данных NCBI, в которой представлены большинство секвенированных на настоящий момент геномов. Работа с поисковой системой проста и удобна: вводим в строку поиска название гена, результат получаем разделенный по группам (статьи, последовательности генов, белков, геномов, структура белков и многое другое), выбираем раздел «гены» (рисунок 1), в этом разделе выбираем ген ND6 нужного нам организма (Homo sapiens).
Попадаем на страницу с различной информацией об этом гене (рисунок 2), где в разделе Genomic sequence, перейдя по ссылке Fasta или GeneBank, мы попадаем на страницу (рисунок 3), где представлена нуклеотидная последовательность искомого гена. Важным положительным моментом является то, что данная поисковая система предоставляет возможность изменения координат изображаемого геномного региона. Благодаря этому мы получили последовательность не только самого гена ND6, но и прилегающих к нему с двух сторон участков длиной 100 нуклеотидов, что необходимо для разработки маркеров.
Аминокислотную последовательность белка ND6 довольно легко можно найти в базе Mitomap – в ней после нуклеотидной последовательности всего митохондриального генома приведены координаты и аминокислотные последовательности всех митохондриальных генов.
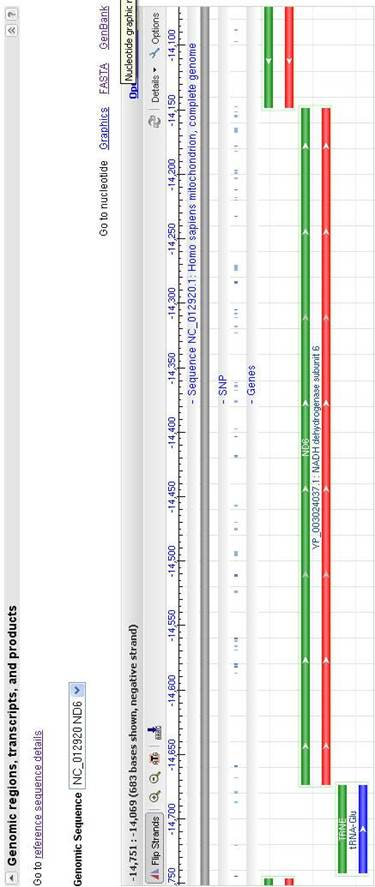
Рисунок 2 – Изображение участка митохондриального генома человека, содержащего ген ND6.
Таким образом, хотя для работы с митохондриальными генами наиболее полная информация представлена в специализированной базе Mitomap, механизмы поиска в ней требуют улучшения, в частности, в ней не хватает инструмента для поиска нуклеотидной последовательности гена по его названию.
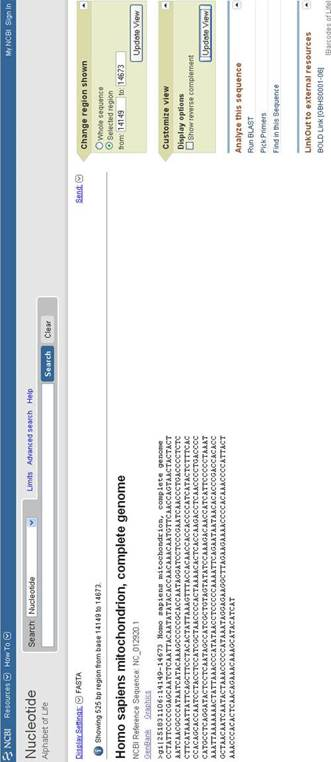
Рисунок 3 – Нуклеотидная последовательность гена ND6.
Подбор праймеров для амплификации гена ND6
Для подбора праймеров к данной нуклеотидной последовательности существует множество программ, в том числе и доступных он-лайн. Для решения нашей задачи мы использовали программу Primer3. Все, что требуется для работы с программой – вставить нуклеотидную последовательность и выделить квадратными скобками ту ее часть, которая является целевой для амплификации. Далее выбираем необходимые параметры праймеров (длина, температура отжига, содержание Г и Ц нуклеотидов, участки матрицы, к которой запрещен подбор праймеров, допустимый уровень самокомплементарности и т.д.) и на выходе получаем наилучшую из пар праймеров, соответствующих данным условиям, а также длину амплифицируемого фрагмента (рисунок 4).
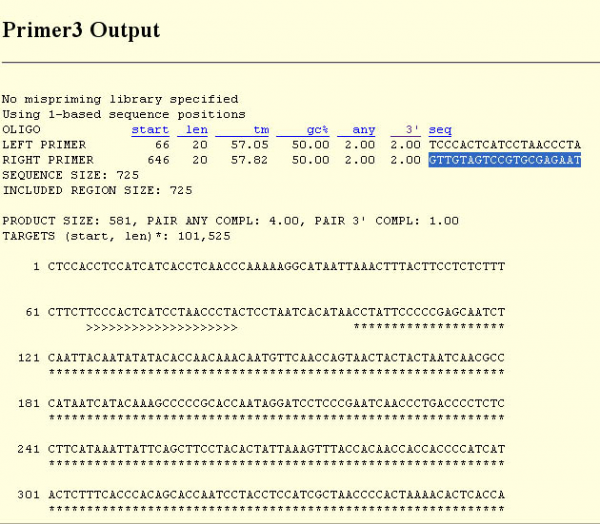
Рисунок 4 – Результат подбора праймеров к гену ND6 с помощью программы Primer3
Далее полученную пару праймеров мы проверили с помощью другой программы (Oligoanalyzer на сайте eu.idtdna.com) для того, чтобы оценить их качество по различным параметрам. Эта программа позволяет определить температуру плавления пары праймер-матрица, стабильность (выраженную в изменении стандартной энергии Гиббса) шпилечных, гомо- и гетеродимерных структур, образуемых данным праймером и их специфичность путем выравнивания с различными геномами с использованием алгоритма BLAST (рисунок 5).
Важным преимуществом этой программы перед некоторыми аналогами является то, что вводными данными являются не только последовательность праймеров, но и концентрации других компонентов ПЦР-смеси (в некоторых других программах для расчетов используются стандартные концентрации этих компонентов, и они не могут быть изменены пользователем).
В результате проверки обоих праймеров с помощью этой программы было показано, что ни один из них не образует стабильных шпилечных или гомодимерных структур, образующийся гетеродимер также не является стабильным при температуре плавления праймеров, которая оказалась равной температуре, рассчитанной программой Primer3. Более того, оба праймера оказались специфичными в отношении гена ND6 именно человека.
Обе программы хорошо подходят для разработки или оценки качества праймеров. Главное преимущество Primer3 заключается в том, что она автоматически подбирает праймеры для данного участка ДНК, тогда как различные олигоаналайзеры, в том числе и рассмотренный здесь, позволяют только проверить соответствие праймеров определенным требованиям, тогда как выбор праймеров необходимо проводить вручную. Однако стоит отметить, что при некоторых специфических задачах без ручного подбора праймеров обойтись нельзя
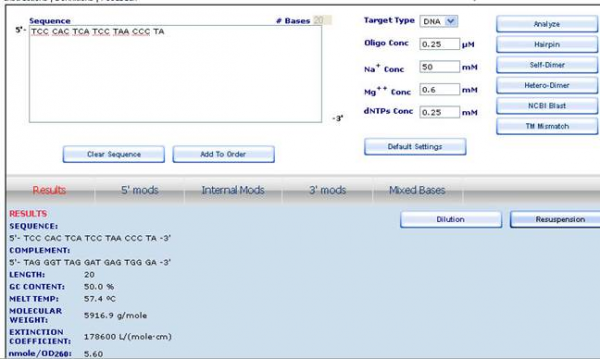
Рисунок 5 Результат анализа праймера с помощью программы Oligoanalyzer.
Сравнение полученной последовательности со стандартной
После амплификации и секвенирования гена ND6 из образца ДНК больного Х, мы получили его последовательность в виде файла хроматограммы, в котором пики уже обработаны, и по ним определена нуклеотидная последовательность. Необходимо определить, имеет ли этот ген какие-либо замены по сравнению с кэмбриджской референсной последовательностью (rCRS) митохондриальной ДНК, принятой исследователями в этой области в качестве стандартной. Для этого мы загружаем rCRS с сайта mitomap.org в виде текстового файла, а потом открываем ее и файл с хроматограммой в программе ChromasPro. Кроме того, что эта программа может воспринимать данные о нуклеотидных последовательностях в разных форматах, она предоставляет ряд возможностей работы с этими последовательностями, в первую очередь выравнивание, в том числе и множественное, и поиск несоответствий в полученном выравнивании, а также поиск открытых рамок считывания, рестрикционных сайтов, трансляция нуклеотидной последовательности в аминокислотную и редактирование нуклеотидных последовательностей в режиме хроматограммы. С помощью ChromasPro мы нашли, что последовательность гена ND6 у больного Х отличается от rCRS по двум позициям: 14456 и 14582 (в обоих случаях выявлена замена А на Г) (рисунок 6).
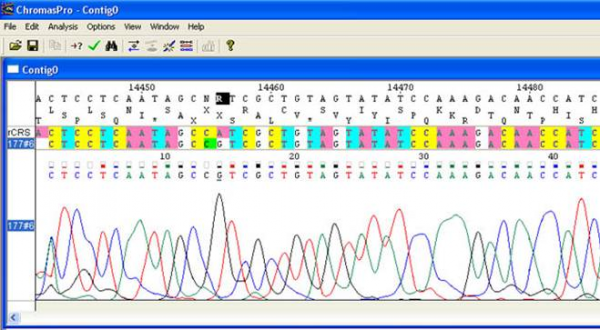
Рисунок 6 – Результат сравнения последовательности гена ND6 больного Х и rCRS. На консенсусной последовательности, приведенной вверху, темным выделен нуклеотид, по которому сравниваемые последовательности различаются.
Основным достоинством программы ChromasPro, наряду с многофункциональностью, является ее удобный, интуитивно понятный интерфейс. Для пользования ей практически не требуется обучение или опыт работы с аналогичными программами.
Выявление функциональной значимости найденных замен
На следующем этапе мы определяли, могут ли обнаруженные замены приводить к нарушению функционирования белка ND6 и, соответственно, вызывать наблюдаемые у больного симптомы. Сначала был проведен поиск этих замен в базе данных Mitomap, в которой собраны все описанные мутации (замены, приводящие к нарушению функции) и полиморфизмы (замены, не сказывающиеся на активности того или иного белка).
Среди описанных мутаций ни одна из двух замен не была найдена, а в списке полиморфизмов была обнаружена замена А-14582-Г. Следовательно, она не может являться причиной наблюдаемой клинической картины.
Для того, чтобы получить представление о возможной роли замены А-14456-Г в возникновении заболевания, мы сначала определили, приводит ли эта нуклеотидная замена к изменению аминокислоты в белке. Для этого использовалась он-лайн программа Virtual Ribosome (виртуальная рибосома).
Важным преимуществом этой программы является то, что она может транслировать ДНК не только в соответствии с классическим генетическим кодом, но и в соответствии с 22 отличных от него вариантов генетического кода. Так как мы работали с митохондриальным геномом, то эта функция нам была необходима, ведь в этом случае мы имеем дело с неканоническим генетическим кодом. В результате мы получаем аминокислотную последовательность белка в однобуквенном формате. Далее, чтобы определить, имеются ли отличия в последовательности аминокислот между «нормальным» белком ND6 и тем белком, который образуется в организме больного Х, мы провели выравнивание этих двух аминокислотных последовательностей с помощью программы SIM, в которую просто вводятся две аминокислотные последовательности, требующие выравнивания. В результате программа выдает две выровненные последовательности, при этом позиции, одинаковые в обоих из них, помечаются знаком «*» (рисунок 8).
В нашем случае белки отличаются по 2 позициям (рисунок): V31A и M73T. Первое отличие вызвано полиморфизмом А-14582-Г, о котором уже шла речь. Вторая замена – как раз та, функцию которой мы определяли с использованием программы SNAP [2].
Говоря о программе SIM, стоит отметить одно ее существенное неудобство – координаты позиций, по которым сравниваемые последовательности приходится отсчитывать вручную, так как никакого способа получить ее автоматически программа не предоставляет.

Рисунок 7 – Результат выравнивания двум аминокислотных последовательностей в программе SIM.
В качестве входных данных программой SNAP используются аминокислотная последовательность «нормального» белка (в нашем случае – белка, соответствующего rCRS) и аминокислотные замены в формате XposY, где X – аминокислота в «нормальном» варианте белка, pos – ее порядковый номер в последовательности и – аминокислота, на которую она заменилась в результате мутации (в нашем случае мы ввели в программу обе замены V31A и M73T, чтобы заодно проверить программу, предложив ей заведомо нейтральную замену).
При предсказании последствий аминокислотной замены для функционирования белка программа основывается на смоделированной на основе его аминокислотной последовательности вторичной структуре, доступности данного участка белковой молекулы для растворителя и степени сходства между двумя аминокислотами. Особенностью программы SNAP является то, что результат она выдает не в окне браузера, а отправляет на введенный пользователем адрес электронной почты. Это связано со значительным временем, необходимым для проведения анализа, и, таким образом, позволяет, позволяет продолжить работу с браузером, не дожидаясь окончания анализа. К тому же, благодаря этому снижается вероятность случайной потери результатов.

Рисунок 8 – Результат оценки функциональной значимости двух аминокислотных замен в белке ND6 с помощью программы SNAP
Через некоторое время после подачи запроса нам на электронную почту пришел результат анализа, изображенный на рисунке 9. Он содержит следующую информацию: характеристику каждой из замен (нейтральная или нет), а также индекс надежности результата (отражает уровень «уверенности» программы в данном результате») и ожидаемая точность. Сразу стоит отметить, что пояснения касательно того, что представляют собой эти численные параметры, приведены только в статье с описанием программы, но не на сайте [2].
Тем не менее, по результатам SNAP анализа было показано, что замена V31A является нейтральной (что соответствует экспериментальным данным), а замена M73T вызывает нарушение функционирования белка ND6, причем этот результат имеет высокий индекс надежности. Следовательно, можно считать, что именно эта аминокислотная замена является причиной клинических проявлений, наблюдаемых у больного Х.
Заключение
Биоинформатика является молодой, но перспективной областью биологической науки, основной задачей которой является работа с большими массивами молекулярно-генетических данных, а также моделирование и предсказание свойств отдельных биологических молекул и целых биологических систем. Подходы, разработанные для применения в биоинформатических исследованиях, такие как методы работы с геномными базами данных, алгоритмы выравнивания последовательностей и предсказания вторичной структуры молекул нуклеиновых кислот и белков и их термодинамических параметров, могут применяться не только в биоинформатических ислледованиях, но и быть полезными в лабораторной молекулярно-генетической работе. В частности, подобные подходы были использованы нами в работе по поиску и функциональной характеристике мутаций в гене ND6 митохондриального генома человека. Применение биоинформатических инструментов позволило сравнительно быстро и эффективно найти в базе данных нужную нуклеотидную последовательность, подобрать к ней праймеры, проанализировать полученную в результате секвенирования последовательность и оценить влияние обнаруженных замен на функционирование соответствующего белка. В будущем можно ожидать интенсивного развития БИ, особенно в таких направлениях, как моделирование биологических систем, предсказание функций генов, систематика биологических объектов и выявление механизмов эволюции органического мира.
Список литературы к реферату
[Электронный ресурс]//URL: https://inzhpro.ru/diplomnaya/bioinformatsionnyie-tehnologii/
1. Achuthsankar SN, Computational Biology & Bioinformatics: A Gentle Overview. // Communications of the Computer Society of India, January 2007.
2. Bromberg Y and Rost B, SNAP: predict effect of non-synonymous polymorphisms on function. // Nucleic Acids Research – 2007, Vol. 35, No. 11, 3823–3835.
3. Doolittle RF, The Roots of Bioinformatics in Protein Evolution // 2010 PLoS Comput Biol 6(7)
4. Drubin DA et al., Designing biological systems. // Genes Dev. 2007 21: 242-254
5. Xuhua X, Bioinformatics and the cell. // Springer, 2007, 363
6.
7. http://elementy.ru/lib/430895
Предметный указатель к реферату
B
BLAST
- 8, 22
M
Mitomap
- 2, 19, 20, 21, 23, 24
Б
базы данных
- 2, 5, 6, 7, 19, 20, 21, 24, 26
биоинформатика
- 3, 4, 5, 6, 7, 11, 13, 15, 17, 19, 27
В
выравнивание
- 2, 5, 7, 8, 14, 19, 22, 23, 24, 26
Г
геном
- 6, 8, 9, 14, 18, 19, 20, 21, 26
М
моделирование
- 2, 4, 5, 6, 11, 12, 17, 18, 26
модель
- 10, 17
С
системная биология
- 2, 3, 6, 17
структура
- 2, 10, 11, 12, 13, 15, 18, 21, 26
Ресурсы для поиска биологической информации
http:// www.ncbi.nlm.nih.gov/pubmed
Pubmed является самой популярной интернет базой ссылок на биологическую и медицинскую литературу. Она содержит примерно 20 млн ссылок на статьи из биологических журналов, литературу из национальной медицинской библиотеки США и он-лайн книги. Как правило, для каждой статьи (книги) приводится название, фамилии авторов, год публикации, название журнала и издательства, аннотация и ссылка на сайт издательства, по которой можно найти электронную версию статьи или книги. Кроме того, для каждой статьи эта база предлагает список статей сходной тематики или содержащие ссылку на рассматриваемую статью.
Pubmed позволяет осуществлять поиск по ключевым словам, имени и инициалам одного или нескольких авторов, году публикации, названию журнала, а также по комбинации этих характеристик.
Pubmed предоставляет пользователям и ряд дополнительных возможностей. Перейдя по ссылке My NCBI, можно создать свой аккаунт, в котором можно завести и редактировать свою коллекцию ссылок, а также сохранять поиск с возможностью получать на электронную почту обновление его результатов. С помощью Pubmed можно сохранять ссылки в формате, позволяющем импортировать их в программы управления библиографической информацией (например, Bibus, JabRef, CiteULike и др.).
Кроме того, имеется текстовое и анимированное руководство по пользованию базой.
Основным неудобством Pubmed является то, что, в базе представлены только англоязычные источники или те, которые имеют перевод названия и резюме на английский.
http:// blast.ncbi.nlm.nih.gov/Blast.cgi
BLAST (Basic Local Alighnment Search Tool) − это еще одна поисковая система, предоставляемая сайтом национального центра биотехнологической информации NCBI. База содержит нуклеотидные последовательности 1400 прокариотических геномов и 225 геном эукариот. С помощью BLAST можно осуществлять поиск конкретной нуклеотидной последовательности во всех или в выбранных геномах, представленных в базе. В таком случае пользователь получает информацию о том, в геноме каких организмов встречается искомая или схожая с ней последовательность, на какой хромосоме, в каком гене или межгенном участке она находится и каковы ее координаты, а также степень соответствия искомой и найденной последовательности. Также BLAST позволяет искать белковые молекулы по последовательности аминокислот или нуклеотидов и нуклеотидные последовательности (гены) по аминокислотной последовательности.
Кроме поиска нуклеотидных или аминокислотных последовательностей, BLAST предоставляет возможности для разработки ПЦР праймеров для определенной последовательности нуклеотидов; выравнивания 2х и более нуклеотидных последовательностей; поиска консервативных доменов в нуклеотидных последовательновтях и других операций.
Хотя для поиска последовательностей BLAST является одной из самых удобных и популярных он-лайн баз, дальнейший анализ последовательностей лучше осуществлять в геномных браузерах, так как они представляют последовательности в более наглядном, удобном и информативном виде (например, трудно выделить экзоны или какие-нибудь регуляторные элементы в последовательности гена).
http:// www.ncbi.nlm.nih.gov/sites/gquery
Entrez cross-database search – объединяет в себе возможности сразу нескольких поисковых систем, в том числе и двух описанных выше. Используя ключевые слова (например, название гена, вид организма, функция или заболевание), можно соответствующие литературные источники, связанные с ключевыми словами нуклеотидные последовательности ДНК и РНК, аминокислотные последовательности и 3D-структуру белковых молекул, возможные вариации в соответствующих генах и много другой полезной информации. Из недостатков данной поисковой системы можно отметить то же, что и для BLAST – некоторое неудобство работы с нуклеотидными последовательностями.
http:// www.mitomap.org/MITOMAP
Mitomap – специализированная база данных для работы с митохондриальным геномом человека. Позволяет просмотреть как весь митохондриальный геном целиком, так и любой его ген или некодирующий участок, аминокислотную последовательность того или иного белка, загрузить последовательность митохондриальной ДНК для работы в различных программах. Кроме того, с помощью Mitomap можно обнаружить описанные мутации различных типов и точки полиморфизма (и определить соответствующую им гаплогруппу) в любом участке митохондриального генома. Эта база является очень удобной в использовании и позволяет получить практически любую информацию, необходимую тем, кто работает с митохондриальной ДНК человека.
Ресурсы для работы с нуклеотидными и аминокислотными последовательностями
http:// www.bioinformatics.org/sms2/
Sequence Manipulation Suite – интернет ресурс, позволяющий осуществлять различные операции с нуклеотидными и аминокислотными последовательностями. Например, пользуясь этим сайтом, можно изменять формат нуклеотидной последовательности, избавляться от символов, не встречающихся в ДНК и белках, разделять последовательности ДНК на триплеты, получать комлементарные молекулы ДНК, переводить однобуквенные обозначения аминокислот в трехбуквенные и обратно.
С помощью этого ресурса можно также выравнивать нуклеотидные и аминокислотные последовательности, переводить нуклеотидную последовательность в аминокислотную и наоборот, искать определенные домены, в том числе CpG островки и открытые рамки считывания в ДНК. Sequence Manipulation Suite предоставляет возможности вычисления молекулярной массы и изоэлектрической точки белковых молекул, построения рестрикционных карт молекул ДНК и разработки ПЦР праймеров. Этот сайт будет полезен любому молекулярному биологу для планирования экспериментов и простой обработки результатов. Главное преимущество ресурса – его многофункциональность. Однако для многих из предоставляемых функций существуют другие, более совершенные программы с большими возможностями.
http://expasy.org/tools/
ExPASy Proteomic tools – самое полное (из известных мне) собрание компьютерных инструментов для анализа аминокислотных последовательностей и протеомных данных, созданное Швейцарским Институтом Биоинформатики (SIB).
Кроме программ, разработанных в самом SIB и хранящихся на сервере института, на сайте представлены и ссылки на программы других разработчиков. Здесь можно найти инструменты для анализа данных масс-спектрометрии и двумерного электрофореза, определения белка по аминокислотной последовательности, молекулярной массе и изоэлектрической точке, расчета молекулярной массы и изоэлектрической точки белка по его аминокислотной последовательности, в том числе с учетом посттрансляционных модификаций. Есть также группа программ для перевода нуклеотидной последовательности в аминокислотную и обратно; поиска сходства между определенной последовательностью и последовательностями, представленными в базах данных; предсказания вторичной, третичной и четвертичной структуры, сигналов внутриклеточной локализации, сайтов посттрансляционных модификаций белков; визуализации третичной и четвертичной структуры белков, сравнения аминокислотной последовательности и третичной структуры двух или более белковых молекул и многого другого.
Важной положительной характеристикой сайта является то, что представленные программы разделены на группы, в зависимости от выполняемых ими функций. Кроме того, рядом с каждой гиперссылкой есть краткое описание программы и области ее применения.
Таким образом, данный ресурс предоставляет максимально широкие возможности работы с белковыми молекулами. Этот сайт будет полезен практически любому биологу, занимающемуся молекулярно-биологическими исследованиями.
Primer3, по моему мнению, является наиболее удобной он-лайн программой для разработки ПЦР-праймеров к определенной последовательности ДНК. Она предлагает пары праймеров, удовлетворяющие параметрам, заданным пользователем. К числу варьируемых параметров относятся длина праймером, температура отжига, их расположение на введенной последовательности ДНК, размер ПЦР-продукта, содержание Г-Ц пар, степень самокоплементарности и комплементарности праймеров друг другу. Программа позволяет быстро и качественно подбирать праймеры для амплификации нужного участка ДНК.
http://vasilipankratov.narod.ru/
магистранта биологического факультета Панкратова В.С.
специальность биология
|
Смежные специальности
|
Основная специальность
|
Сопутствующие специальности нет |
Вопрос по специальности
<questiongroup groupname=»122BF-2″ no=»122″ mark=»1″ amountrate=»1″>
- <questions>
- <question type=»sequence»>
- <text>В какой последовательности нужно выполнять данные действия при поиске патогенной мутации в конкретном гене?</text>
- <stoptime>false</stoptime>
- <attachfile>false</attachfile>
- <answers type=»request»>
- <answer num=»5″ right=»1″>Проверить наличие найденных замен в соответствующей базе данных</answer>
- <answer num=»7″ right=»1″>сделать вывод о наличии патогенных замен в изучаемом гене</answer>
- <answer num=»2″ right=»1″>подобрать праймеры для амплификации изучаемого гена</answer>
- <answer num=»1″ right=»1″>найти последовательность нужного гена в геномной базе данных</answer>
- <answer num=»3″ right=»1″>провести амплификацию и секвенирование</answer>
- <answer num=»6″ right=»1″>если найденная замена не описана — оценить ее влияние на функцию белка с помощью специального ПО</answer>
- <answer num=»4″ right=»1″>сравнить полученную последовательность с эталонной</answer>
- </answers>
- </question>
- </questions>
- </questiongroup>
Вопрос по общему курсу
<questiongroup groupname=»122BF-2″ no=»622″ mark=»1″ amountrate=»1″>
- <questions>
- <question type=»correspondence»>
- <text>Для каждого атрибута тега body подберите соответствующее свойство из языка CSS</text>
- <stoptime>false</stoptime>
- <attachfile>false</attachfile>
- <answers type=»request»>
- <set setsid=»1″ setnumber=»1″ no=»2″>bgcolor</set>
- <set setsid=»2″ setnumber=»1″ no=»1″>vlink</set>
- <set setsid=»3″ setnumber=»1″ no=»4″>text</set>
- <set setsid=»4″ setnumber=»1″ no=»3″>alink</set>
- <set setsid=»1″ setnumber=»2″ no=»1″>background</set>
- <set setsid=»2″ setnumber=»2″ no=»2″>:visited</set>
- <set setsid=»3″ setnumber=»2″ no=»3″>color</set>
- <set setsid=»4″ setnumber=»2″ no=»4″>:active</set>
- <reference setsfrom=»1″ setsto=»1″/>
- <reference setsfrom=»2″ setsto=»2″/>
- <reference setsfrom=»3″ setsto=»3″/>
- <reference setsfrom=»4″ setsto=»4″/>
- </answers>
- </question>
- </questions>
- </questiongroup>
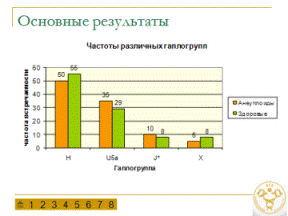
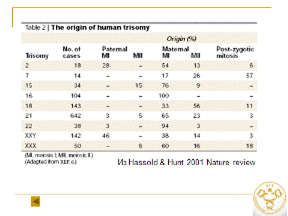
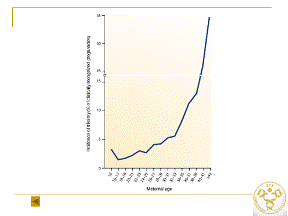
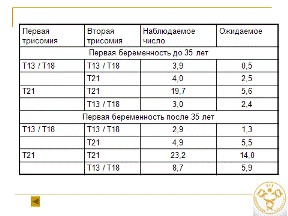
Особенности митохондриального генома у больных анеуплоидиями
1. Access 2000: самоучитель / П.Ю.Дубнев. – М.: ДМК Пресс, 2004. – 313 с., ил.
2. Access 2000: справочник / Гюнтер Штайнер; науч. ред. С.И.Молявко. – М.: Лаборатория Базовых Знаний, 2000. – 474 с.
3. Excel 2000 / Марк Зайден; науч. ред. А.Плещ, С.Молявко. – М.: Лабо-ратория Базовых Знаний, 1999. – 328 с., ил.
4. Microsoft Office 2000 / Стив Сагман; пер. с англ. А.И.Осипова, П.А.Мерещук. – М.: ДМК, 2002. — 669 c., ил.
5. Microsoft Office XP в целом: наиб. полное рук-во. Для широкого круга пользователей / Ф.Новиков, А.Яценко. – Спб: БХВ-Петербург, 2002. – 917 с., ил.
6. Microsoft PowerPoint 2003: самоучитель / М.В.Спека. – Москва, Санкт-Петербург, Киев: Диалектика, 2004. – 363 с., ил.
7. Microsoft Word 2003 в теории и на практике / С.Бондаренко, М.Бондаренко. – Минск: Новое знание, 2004. – 336 с., ил.
8. Windows 2000: проблемы и решения. Спец. справочник / Мэтью Штре-бе; пер. с англ.П.Анджан, А.Войтенко. – Спб: Питер, 2002 – 858 с.
9. Word 2000 / Марк Зайден; науч. ред. В.Гребнев, С.Молявко. – М.: Ла-боратория Базовых Знаний, 1999. – 336с., ил.
10. Ваш Office 2000: MS Word, MS Excel, Internet Explorer и др. / С.Баричев, О.Плотников. – М.: КУДИЦ ОБРАЗ, 2000. – 318 с., ил.
11. Информатика: учеб. для студ. вузов, обуч. по естеств.-науч. напр. и спец. / В.А.Каймин. – М.: ИНФРА-М, 2000. – 232 с., ил.
12. Компьютерные презентации: от риторики до слайд-шоу / Т.М.Елизаветина. – М.: КУДИЦ ОБРАЗ, 2003. – 234 с.
13. Освой самостоятельно Microsoft Excel 2000 = Teach Yourself Microsoft Excel 2000: 10 минут на урок: учеб. пособие: пер. с англ. / Дженнифер Фултон – М.: Издательский дом «Вильямс», 2000. – 224 с., ил.
14. Технологии работы с текстами и электронными таблицами: Word, Excel / М.С.Шибут; под ред. И.Ф.Богдановой. Минск: общественное объеди-нение «Молодежное научное общество», 2000. – 142 с.
15. Шафрин Ю.А. Информационные технологии: учеб. пособие: В 2 ч / Ю.А.Шафрин. – М.: Лаборатория Базовых Знаний, 2003. – Ч.1: Основы информатики и ИТ – 316 с., ил.
16. Шафрин Ю.А. Информационные технологии: учеб. пособие: В 2 ч / Ю.А.Шафрин. – М.: Лаборатория Базовых Знаний, 2003. – Ч.1: Офис-ная технология и ИТ – 336 с., ил.