
Вещество, на поверхности которого происходит А., называется адсорбентом, а поглощаемое из объёмной фазы — адсорбатом. В зависимости от характера взаимодействия между молекулой адсорбата и адсорбентом А. принято подразделять на физическую А. и хемосорбцию. Менее прочная физическая А. не сопровождается существенными изменениями молекул адсорбата. Она обусловлена силами межмолекулярного взаимодействия, которые связывают молекулы в жидкостях и некоторых кристаллах и проявляются в поведении сильно сжатых газов. При хемосорбции молекулы адсорбата и адсорбента образуют химические соединения. Часто А. обусловлена и физическими и химическими силами, поэтому не существует чёткой границы между физикой А. и хемосорбцией.
Хемосорбция
Адсорбированные молекулы не только совершают движение вдоль поверхности адсорбента, но и колеблются, то приближаясь к поверхности, то удаляясь от неё. Чем выше температура, тем интенсивнее колебательное движение, а стало быть, больше вероятность того, что в процессе таких колебаний связь молекулы с поверхностью будет разорвана и молекула десорбируется. Благодаря этому с ростом температуры уменьшается время А. и равновесное количество адсорбированных молекул.
С ростом концентрации или давления адсорбата в объёме увеличивается частота попаданий молекул адсорбата на поверхность адсорбента; пропорционально ей возрастает скорость А. и увеличивается равновесное количество адсорбированных молекул. Кривые зависимости равновесной А. от концентрации или давления адсорбата при постоянной температуре называются изотермами А.
Если адсорбат покрывает поверхность слоем толщиной в одну молекулу, А. называется мономолекулярной. Простейшая изотерма мономолекулярной А. представляет собой прямую линию, выходящую из начала координат, где на оси абсцисс отложено давление адсорбата Р, а на оси ординат степень заполнения поверхности Q, т. е. доля поверхности, покрытая адсорбированными молекулами. Это — т. н. изотерма Генри:
Q = kP.
Коэффициент пропорциональности k зависит главным образом от температуры и характера взаимодействия адсорбент — адсорбат.
Уравнение Генри справедливо при очень низких степенях заполнения для однородной поверхности. По мере увеличения степени заполнения всё большую роль начинает играть взаимодействие между адсорбированными молекулами и интенсивность их поверхностной подвижности. Если молекулы адсорбата притягиваются друг к другу, то каждая вновь адсорбирующаяся молекула будет испытывать притяжение и адсорбата и молекул, адсорбированных ранее. Поэтому, по мере заполнения поверхности, силы, удерживающие адсорбированную молекулу, будут увеличиваться и условия для А. будут всё более и более благоприятными. В этом случае с ростом давления изотерма всё круче и круче идёт вверх (см. кривую 1 ).
Давление в жидкости и газе (2)
... поверхность площадью 1см2 за 1 сек. выражается двадцатитрехзначным числом. Хотя сила удара отдельной молекулы мала, но действие всех молекул о стенки сосуда значительно, оно и создает давление газа. Итак, давление газа ... [Электронный ресурс]//URL: https://inzhpro.ru/referat/davlenie-gaza-klass/ 1. Учебники по Физике за 7-9 Классы. 2. Элементарный учебник Физики (том 1-2). 3. Справочник по Физики ...
Однако по мере заполнения поверхности вновь адсорбирующимися молекулами становится всё труднее найти свободное (не занятое др. молекулами адсорбата) место на поверхности. Поэтому с увеличением давления рост А. замедляется и степень покрытия стремится к постоянному значению, равному единице (см.кривую 2, которая характерна при отсутствии взаимного притяжения молекул адсорбата).
Если действуют оба эти фактора, то получаются вогнуто-выпуклые изотермы (см.кривую 3 ).
Выпуклые изотермы (см. кривую 2 ) часто описывают уравнением Ленгмюра
![]()
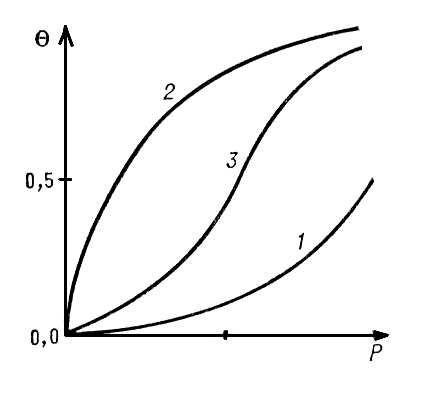
Здесь а — адсорбционный коэффициент, аналогичный по физическому смыслу константе Генри k. Уравнение Ленгмюра справедливо для мономолекулярной А. на однородной поверхности, если можно пренебречь притяжением молекул адсорбата между собой и их подвижностью вдоль поверхности.
Смачивание
ккал/моль,
А. играет важную роль при теплообмене между газообразными, жидкими и твёрдыми телами. например, молекулы газа, адсорбируясь на горячей поверхности, приобретают энергию, соответствующую температуре поверхности, и после десорбции сообщают эту энергию другим молекулам газа, нагревая его. Это не единственный, но важный механизм теплообмена.
Дисперсные системы, Мицелла, Коагуляция
При крашении тканей, в полиграфической промышленности имеют дело с А. молекул красителей. При производстве полимеров наполнителями служат адсорбенты. В вакуумной технике А. на стенках откачиваемой аппаратуры замедляет скорость откачки и ухудшает вакуум, однако, с другой стороны, действие различных сорбционных насосов основано на явлении А. В радиоэлектронной промышленности А. используется для стабилизации электрических свойств полупроводниковых приборов. Вообще во всех явлениях и процессах, где существенны поверхностные свойства, А. играет важную роль.
экстракцией.
абсорбер.
сорбентом,
Поверхностное натяжение, важнейшая термодинамическая характеристика поверхности раздела фаз (тел), определяемая как работа обратимого изотермического образования единицы площади этой поверхности. В случае жидкой поверхности раздела П. н. правомерно также рассматривать как силу, действующую на единицу длины контура поверхности и стремящуюся сократить поверхность до минимума при заданных объёмах фаз. Применительно к легкоподвижным поверхностям оба определения равнозначны, но первое предпочтительнее, т.к. имеет более ясный физический смысл. П. н. на границе двух конденсированных фаз обычно называется межфазным натяжением. Работа образования новой поверхности затрачивается на преодоление сил межмолекулярного сцепления ( когезии ) при переходе молекул вещества из объёма тела в поверхностный слой. Равнодействующая межмолекулярных сил в поверхностном слое не равна нулю (как в объёме тела) и направлена внутрь фазы с большей когезией. Таким образом, П. н. — мера некомпенсированности межмолекулярных сил в поверхностном (межфазном) слое или, что то же, избытка свободной энергии в поверхностном слое по сравнению со свободной энергией в объёмах соприкасающихся фаз. В соответствии с определениями П. н. его выражают в дж/м 2 или н/м (эрг/см 2 или дин/см ).
Измерение динамической вязкости жидкостей и газов
... молекулы «нижнего» слоя, и поэтому ускоряет нижний слой. Δр х /Δ 2. Определение вязкости воздуха по методу Пуазейля 2.1. Теория метода При ламинарном движении жидкостей и газов ... называемой кинематической вязкостью, равной динамической вязкости жидкости, деленной на плотность жидкости . временем «оседлой жизни» молекулы. «оседлой жизни» 1.2. Движение твердого тела в жидкости ламинарное течение, ...
поверхностной энергии.
В общем случае многокомпонентных систем в соответствии с термодинамическим уравнением Гиббса при адсорбции изменение П. н.
- d s = Г1 d m1 + Г2 d m2 +…,
где Г 1 , Г2 ,… — поверхностные избытки компонентов 1, 2,…, т. е. разность их концентраций в поверхностном слое и объёме раствора (или газа), a d m1 , d m2 ,… — изменения химических потенциалов соответствующих компонентов (знак «минус» показывает, что П. н. при положительной адсорбции уменьшается).
Разницей в П. н. чистой жидкости и жидкости, покрытой адсорбционным монослоем, определяется поверхностное давление.
анизотропии
Л. А. Шиц.
адсорбцию
Смачивание, явление, возникающее при соприкосновении жидкости с поверхностью твёрдого тела или другие жидкости. Оно выражается, в частности, в растекании жидкости по твёрдой поверхности, находящейся в контакте с газом (паром) или другой жидкостью, пропитывании пористых тел и порошков, искривлении поверхности жидкости у поверхности твёрдого тела. Так, С. вызывает образование сферического мениска в капиллярной трубке, определяет форму капли на твёрдой поверхности или форму газового пузырька, прилипшего к поверхности погруженного в жидкость тела. С. часто рассматривают как результат межмолекулярного (вандерваальсова) взаимодействия в зоне контакта трёх фаз (тел, сред).
Однако во многих случаях, например при соприкосновении жидких металлов с твёрдыми металлами, окислами, алмазом, графитом, С. обусловлено не столько межмолекулярным взаимодействием, сколько образованием химических соединений, твёрдых и жидких растворов, диффузионными процессами в поверхностном слое смачиваемого тела. Тепловой эффект, сопровождающий соприкосновение жидкости со смачиваемой поверхностью, называется теплотой смачивания.
Мерой С. обычно служит краевой угол q между смачиваемой поверхностью и поверхностью жидкости на периметре С. ( рис. 1 ).
Угол q отсчитывают со стороны жидкости. При статическом (равновесном) С. он связан с поверхностным натяжением жидкости (sж ), поверхностным натяжением твёрдого тела (sт ) и межфазным натяжением на границе твёрдое тело — жидкость (sтж ) уравнением Юнга: cosq = (sт — sтж )/ (ж . Величиной угла q оценивают лиофильность илиофобность поверхностей по отношению к различным жидкостям. На лиофильной поверхности жидкость растекается, т. е. имеет место частичное (0° 90°) (см. рис. 2 ).
Краевой угол зависит от соотношения сил сцепления молекул жидкости с молекулами или атомами смачиваемого тела (адгезия )и сил сцепления молекул жидкости между собой (когезия ). Обратимую работу адгезии и когезии вычисляют соответственно по уравнениям: Wa =sж (1 + cosq) и W k = 2sж . При Wa K всегда q>0°, причём с увеличением отношения Wa k улучшается С. Разность S = W a /W k называется коэффициентом растекания. Часто наблюдаемая задержка в установлении равновесных краевых углов называется гистерезисом С. Различают кинетический (динамический) и статический гистерезис С. Причиной гистерезиса может быть шероховатость поверхности, особенности структуры поверхностного слоя, релаксационные процессы в жидкой фазе и др. Если твёрдое тело соприкасается одновременно с двумя несмешивающимися жидкостями, происходит избирательное С. Эффективные регуляторы С. — поверхностно-активные вещества, которые могут как улучшать, так и ухудшать С.
Моющее действие
смачивание
дисперсности
поверхностное натяжение
Д. с. могут быть бесструктурными (свободнодисперсными) и структурированными (связнодисперсными).
Структурированные Д. с. пронизаны сеткой-каркасом из соединённых между собой частиц (капель, пузырьков) дисперсной фазы, вследствие чего обладают некоторыми механическими свойствами твёрдых тел (подробнее см. Дисперсная Гели).
Характерная особенность Д. с. — высокая свободная энергия как следствие сильно развитой межфазной поверхности; поэтому Д. с. обычно (кроме лиофильных Д. с.) термодинамически неустойчивы. Они обладают повышенной адсорбционной способностью, химической, а иногда и биологической активностью. Д. с. — основной объект изучения коллоидной химии.
Д. с. широко распространены в природе, технике и быту. Примерами Д. с. могут служить горные породы, грунты, почвы, дымы, облака, атмосферные осадки, растительные и животные ткани; строительные материалы, краски, моющие средства, волокнистые изделия, важнейшие пищевые продукты и многие др.
Классификация дисперсных систем по агрегатному состоянию фаз
|
Дисперсионная среда |
Дисперсная фаза |
||
|
газовая |
жидкая |
твёрдая |
|
|
Газовая |
Дисперсные систе- мы не образуются |
Туманы |
Дымы, пыль |
|
Жидкая |
Пены |
Эмульсии |
Суспензии |
|
Золи (коллоидные «растворы»)* |
|||
|
Твёрдая |
Аэрогели (пористые тела) |
Жидкие включения в твёрдых телах |
Твёрдые золи (рубиновое стекло) |
* Предельно высокодисперсные системы (золи) иногда трудно классифицировать по агрегатному состоянию дисперсной фазы.
Сольватация
броуновского движения
В лиофильных золях, коллоидных дисперсиях типа гидрозолей мыл, например олеата натрия или лаурилсульфата калия, М. представляет собой ассоциат (объединение) молекул. В каждой такой молекуле длинный углеводородный (гидрофобный) радикал связан с полярной (гидрофильной) группой. При образовании М. несколько десятков или сотен молекул объединяются так, что гидрофобные радикалы образуют ядро (внутреннюю область), а гидрофильные группы — поверхностный слой М. Если дисперсионной средой является органическая жидкость, ориентация молекул в М. может быть обратной: в ядре сосредоточатся полярные группы, тогда как гидрофобные радикалы будут обращены во внешнюю фазу. Изобразив молекулу мицеллообразующего вещества в виде волнистой линии (гидрофобный радикал) с кружочком на конце (гидрофильная группа), можно представить простейшие структурные типы М. схемами:
![]()
Полуколлоидные системы
Наличием М. объясняется моющее действие водных растворов (точнее, коллоидных дисперсий) мыл, а также некоторые явления в биологических системах и при технологических процессах (см. также Солюбилизация).
Коагуляция (от лат. Coagulatio — свёртывание, сгущение), слипание частиц коллоидной системы при их столкновениях в процессе теплового (броуновского) движения, перемешивания или направленного перемещения во внешнем силовом поле. В результате К. образуются агрегаты — более крупные (вторичные) частицы, состоящие из скопления более мелких (первичных).
Первичные частицы в таких скоплениях соединены силами межмолекулярного взаимодействия непосредственно или через прослойку окружающей (дисперсионной) среды. К. сопровождается прогрессирующим укрупнением частиц (увеличением размера и массы агрегатов) и уменьшением их числа в объёме дисперсионной среды — жидкости или газа.
Различают быструю и медленную К. При быстрой К. почти каждое соударение частиц эффективно, т. е. приводит к их соединению; при медленной К. соединяется часть сталкивающихся частиц. В жидкой среде, например при К. золей , укрупнение частиц до известного предела (приблизительно до размера 10-4 см ) не сопровождается их оседанием или всплыванием. Это скрытая К., при которой система сохраняет седиментационную устойчивость. Дальнейший рост частиц приводит к образованию сгустков или хлопьев (флокул), выпадающих в осадок (коагулят, коагель) или скапливающихся в виде сливок у поверхности; это явная К. В некоторых случаях при К. во всём объёме дисперсионной среды возникает рыхлая пространственная сетка (коагуляционная структура) и расслоения системы не происходит (см. Гели ).
Если коллоидные частицы — капельки жидкости или пузырьки газа, то К. может завершиться их слиянием, коалесценцией .
Коллоидные системы
биополимеров
реакции химические
полупроводниковых материалов
катализаторов
активированного комплекса
К. не связан с изменением свободной энергии катализатора, и воздействие катализатора не может поэтому смещать положение равновесия химической реакции. Вблизи состояния равновесия катализаторы в равной степени ускоряют как прямую, так и обратную реакцию.
Основным фактором, определяющим скорость химического превращения, является энергия активации ( Е )— разность энергий активного комплекса и исходных реагирующих молекул. Если предположить, что реакция не нарушает равновесного распределения энергии между молекулами, то вероятность образования активного комплекса, а следовательно, и скорость реакции в первом приближении пропорциональна exp (—E/RT ), где R — газовая постоянная, Т — абсолютная температура. Отсюда следует, что скорость реакции тем больше, чем меньше Е , и вследствие экспоненциальной зависимости возрастает значительно даже при небольшом снижении Е .
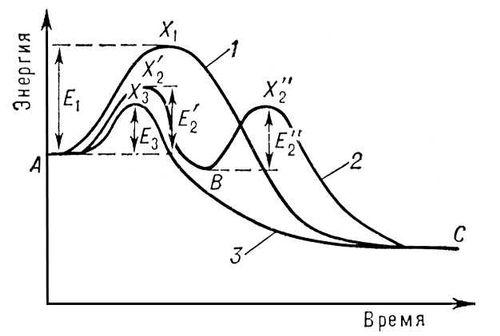
На рис. представлено изменение энергии при реакции без катализатора (кривая 1 ) и при участии катализатора (кривые 2 и 3 ).
Кривая 2 с двумя максимумами соответствует образованию одного промежуточного продукта. Число стадий и промежуточных продуктов часто бывает значительно большим. Взаимодействие реагирующих веществ с катализатором может и не приводить к образованию стабильной формы промежуточного соединения (кривая 3 ).
Но и в этом случае катализатор входит в состав активного комплекса и взаимодействие реагирующих веществ с катализатором определяет реакционный путь. Если энергии активных комплексов всех стадий реакционного пути с участием катализатора ниже энергии активного комплекса реакции без катализатора (т. е. ![]() ,
,![]() и E 3 ниже E 1 ), то участие катализатора приведёт к увеличению скорости (положительный К.).
и E 3 ниже E 1 ), то участие катализатора приведёт к увеличению скорости (положительный К.).
Во многих случаях К. ускорение реакции достигается благодаря появлению богатых энергией частиц в процессе самой реакции, причём их концентрация может превосходить равновесную (см. Цепные реакции ).
Например, каталитическое воздействие воды на окисление окиси углерода связано с развитием реакционных путей с участием гидроксильных групп и атомов водорода. Отрицательный К. часто связан с прекращением цепной реакции вследствие обрыва цепей при взаимодействии отрицательного катализатора с активными частицами. Примером может служить замедляющее влияние кислорода на соединение водорода с хлором.
Характер промежуточного химического взаимодействия при К. весьма разнообразен. Обычно различают две группы каталитических процессов: кислотно-основной (гетеролитический) и окислительно-восстановительный (гомолитический).
В процессах первой группы происходит промежуточное кислотно-основное взаимодействие реагирующих веществ с катализатором, например переход протона от катализатора к реагирующим веществам или наоборот. На последующих стадиях протон перемещается в обратном направлении, и катализатор восстанавливает свой состав. При К. апротонными кислотами взаимодействие осуществляется через свободную пару электронов реагирующего вещества. Примерами кислотно-основного К. могут служить гидролиз сложных эфиров, ускоряемый кислотами; гидратация олефинов в присутствии фосфорно-кислотных катализаторов; изомеризация и крекинг углеводородов на алюмосиликатных катализаторах; алкилирование; полимеризация и многие другие реакции. При реакциях окислительно-восстановительного К. промежуточное взаимодействие связано с электронными переходами между катализатором и реагирующими веществами. К этой группе относятся окисление двуокиси серы в трёхокись в производстве серной кислоты; окисление аммиака до окиси азота при получении азотной кислоты; многочисленные процессы парциального окисления органических соединений, например этилена в окись этилена, нафталина во фталевый ангидрид; гидрогенизация; дегидрогенизация; циклизация и ароматизация углеводородов; разложение перекиси водорода и многие др. Каталитической активностью в отношении окислительно-восстановительных реакций обладают преимущественно металлы 4-, 5- и 6-го периодов системы Д. И. Менделеева, имеющие недостроенную d -оболочку электронов, их соединения и в меньшей мере соединения элементов с достраивающейся f -оболочкой (лантаноиды и актиноиды).
Рассмотренные группы далеко не охватывают всё разнообразие каталитических реакций. Характер промежуточного взаимодействия при К. гораздо более сложен и зависит от всех деталей электронной структуры как реагирующих веществ, так и катализатора. Конкретные механизмы каталитических реакций многообразны и пока лишь в немногих случаях твёрдо установлены.
В зависимости от фазового состояния реагирующих веществ и катализатора различают гомогенный и гетерогенный К. Промежуточное положение занимает микрогетерогенный К. в коллоидных системах (например, К. ферментами).
При гомогенном К. катализатор и реагирующие вещества образуют одну однородную систему, границы раздела между катализатором и реагирующими веществами отсутствуют. При гетерогенном К. катализатор и реагирующие вещества находятся в разных фазах и отделены друг от друга границей раздела. Наиболее важны случаи, когда катализатор является твёрдым телом, а реакционная система образует жидкую или газообразную фазу. Промежуточное взаимодействие происходит при этом преимущественно на поверхности твёрдого катализатора.
Выбор состава катализатора для определённой реакции является очень сложной проблемой, решаемой пока главным образом эмпирическим путём. В СССР предложен и развит ряд теоретических подходов, основанных на корреляции отдельных частных свойств катализаторов с их активностью. Так, мультиплетная теория К. (первые публикации 1929) предполагает промежуточное взаимодействие реагирующих веществ с несколькими атомами на поверхности твёрдых катализаторов и придаёт решающее значение соответствию расстояний между атомами в молекулах реактантов и параметров кристаллической структуры катализатора. В дальнейшем теория была дополнена представлением о необходимости определённого соответствия энергий связей, разрывающихся и образующихся в результате реакции, и энергий связей реактантов с катализатором при промежуточном взаимодействии. Значительное распространение в 50-х гг. получило представление о зависимости каталитической активности твёрдых катализаторов, обладающих полупроводниковыми свойствами, от их электрических характеристик, — так называемая электронная теория К. По этой теории предполагается, что промежуточное взаимодействие реактантов с катализатором осуществляется при участии электронов проводимости твёрдого катализатора и поэтому зависит от его коллективных электронных свойств — расположения энергетических зон и локальных уровней электронов, работы выхода электрона, концентрации носителей тока и др. В гетерогенном К. широко использовалось предположение (выдвинутое в 1939) о существовании на поверхности твёрдых катализаторов особых активных центров, представляющих собой ребра, углы или различные структурные нарушения (дислокации) нормальной кристаллической структуры. Предполагалось также, что при нанесении каталитически активного вещества на инертный носитель особые каталитические свойства проявляют отдельно расположенные атомы или совокупности небольшого числа атомов — ансамбли.
Появление точных методов определения поверхности катализаторов позволило установить, что активность, отнесённая к единице поверхности (удельная каталитическая активность), определяется химическим составом и очень мало зависит от структурных дислокаций. Удельная каталитическая активность различных граней кристаллов иногда различается в несколько раз. Большое влияние на активность оказывают нарушения химического состава (отклонение от стехиометрии, внедрение примесей, локальные химические образования и т.п.).
В 60-е годы промежуточное химическое взаимодействие в гетерогенном К. рассматривается преимущественно как локальное, определяемое электронной структурой отдельных атомов или ионов каталитически активного компонента на поверхности катализатора с учётом влияния ближайшего окружения. Значительную помощь в развитии этого подхода оказала обнаруженная экспериментально аналогия в действии твёрдых катализаторов, содержащих определённый металл, при гетерогенном К. и растворимых комплексов, компонентом которых является тот же металл, при гомогенном К. в растворах. При этом используются теории кристаллического поля и поля лигандов, ещё ранее успешно применявшиеся в химии комплексных соединений. Для ряда классов катализаторов и каталитических реакций установлены корреляции между каталитической активностью и энергиями связей реактантов с катализатором при промежуточном взаимодействии, облегчающие в отдельных случаях подбор катализаторов.
Первые научные сведения о К. относятся к началу 19 в. В 1806 французские химики Н. Клеман и Ш. Дезорм открыли каталитическое действие окислов азота на окисление сернистого газа в камерном процессе получения серной кислоты, В 1811 русский химик К. С. Кирхгоф открыл, что разбавленные кислоты способны вызывать превращение крахмала в сахар (глюкозу); в 1814 им же было установлено, что эту реакцию может катализировать диастаза из ячменного солода, — так было положено начало изучению биологических катализаторов — ферментов. В 1818 французский химик Л. Тенар установил, что большое число твёрдых тел оказывает ускоряющее действие на разложение растворов перекиси водорода, а английский химик Г. Дэви открыл способность паров спирта и эфира окисляться кислородом на платине. В 1822 нем. химик И. Дёберейнер установил, что водород и кислород соединяются на платине при обычной температуре. За этим последовало открытие и ряда др. примеров резкого положительного действия веществ на скорость или возникновение химических реакций. Это привело к выделению особой группы явлений, названных нем. химиком Э. Мичерлихом контактными (1833) и швед. химиком И. Берцелиусом каталитическими (1835).
В дальнейшем было открыто большое число каталитических реакций, и за последние 50 лет К. стал ведущим методом осуществления химических реакций в промышленности. Применение катализаторов позволяет проводить химические превращения с высокими скоростями при небольших температурах — большинство промышленных каталитических процессов без катализаторов вообще не могло бы быть реализовано. Подбирая катализаторы, можно направлять химические превращение в сторону образования определённого продукта из ряда возможных. Применение стереоспецифичных катализаторов позволяет регулировать и строение конечных продуктов, например полимеров. С помощью К. в начале 20 в. была решена проблема фиксации азота воздуха. Промотированные железные и другие катализаторы позволили преодолеть химическую инертность элементарного азота и осуществить синтез аммиака. Одновременно был разработан каталитический метод получения азотной кислоты путём окисления аммиака на платиновых сетках. На каталитических реакциях основываются современные методы получения водорода из природного газа. Каталитические методы занимают господствующее положение и в технологии нефтепереработки. Сотни миллионов тонн высококачественного моторного топлива производятся с помощью каталитических реакций крекинга, гидрокрекинга, риформинга, циклизации и изомеризации углеводородов нефти. Особенно большую роль играют каталитические методы в осуществлении процессов органического синтеза. В нашей стране впервые в мире было разработано и реализовано производство синтетического каучука, основанное на превращении этилового спирта в дивинил с помощью многокомпонентного окисного катализатора Лебедева. Каталитические методы используются для получения подавляющего большинства продуктов нефтехимического синтеза: растворителей, ароматических углеводородов, мономеров для производства синтетических каучуков, синтетических волокон и др. полимерных материалов. Катализаторы широко используются и для полимеризации.
К. играет ведущую роль в химических превращениях в живой природе. Вся сложная система управления жизненными процессами в организмах основана на каталитических реакциях. Биологические катализаторы, называемые ферментами или энзимами, представляют собой вещества белковой природы с химически активными группами, часто включающими в свой состав атомы переходных элементов. По некоторым свойствам ферменты превосходят промышленные катализаторы. В СССР и за рубежом широко ведутся исследования новых типов сложных синтетических катализаторов — комплексных соединений, органических полупроводников, полимеров, характеризующихся более простым составом по сравнению с ферментами, но моделирующих в известной степени их действие. Науке о К. принадлежит существенная роль как в прогрессе химической промышленности, так и в раскрытии важнейших биологических закономерностей.
Список литературы
[Электронный ресурс]//URL: https://inzhpro.ru/referat/adsorbentyi/
Лит.: Курс физической химии, т. 1, М., 1964; Бур Я.Х., Динамический характер адсорбции, пер. с англ., М., 1962; Трепнел Б., Хемосорбция, пер. с англ., М., 1958; Бладергрен В., Физ. химия в медицине и биологии, пер. с нем., М., 1951.