
В настоящее время около 40 % потребляемой в мире энергии производится путем сжигания нефтепродуктов. Ежегодная мировая добыча составляет около 4,3 млрд. т. нефти.
До настоящего дня ведутся споры по поводу запасов нефти и газа, однако все прогнозы относительно истощения месторождений нефти не оправдались. Фактически уровень добычи нефти и газа ежегодно возрастает. Согласно расчетам, основанным на теории абиогенно-мантийного происхождения нефти и газа, запасы нефти практически неисчерпаемы в обозримом будущем, по крайней мере, на ближайшие 100 лет. Человечеству дается возможность пользоваться природными запасами наиболее эффективного источника энергии, однако, поскольку нефть содержит ядовитые ингредиенты, ее добыча и переработка может стать причиной экологической катастрофы. Выбросы в атмосферу предприятиями нефтяной и нефтеперерабатывающей промышленности РФ ежегодно составляют около 3,5 млн.т, в водные источники выбрасывается около 5 млрд. м 3 стоков. Особенно большую опасность несут в себе соединения серы. В среднем количество сернистых соединений, входящих в состав нефти таково, что содержание серы в ней более 1%. Ежегодно извлекается с нефтью не менее 43 млн. т серы. Большинство соединений серы токсичны для теплокровных животных. Кроме того, сгорая, сернистые соединения, входящие в состав нефтяных топлив, образуют сернистый газ, который может быть причиной лейкемии и заболеваний органов дыхания. Окисляясь, сернистый газ в атмосфере образует серную кислоту, которая является причиной кислотных дождей. В отдельных городах страны атмосфера загрязнена отработанными газами двигателей автомототранспорта. Их количество доходит до 70-80% от общего выброса загрязняющих веществ в атмосферу.
Пути улучшения состояния окружающей среды путем сокращения выбросов известны. Это, прежде всего, внедрение новых видов моторного топлива с менее токсичным составом отработавших газов, регулировка двигателей внутреннего сгорания до показателей, установленных ГОСТом на токсичность и дымность.
По этой причине ограничивается содержание серы в нефтепродуктах, в частности моторных топливах и мазуте. Содержание серы в бензине ограничивается стандартами EN, ASTM и ГОСТ РФ величинами 0,050,015 %. Особо отличаются своей токсичностью меркаптаны, их количество в бензине ограничивается концентрацией 0,001%. Кроме вредоносного действия на живые организмы, меркаптаны являются сильными коррозионными агентами. В связи с высокой коррозионной активностью содержание меркаптанов в газах и газовых конденсатах, подлежащих транспортировке в трубопроводах, не должно превышать 0,036 % как в умеренном климатическом районе, так и холодном.
Каталог :: Химия. Доклад про нефть
... каждого и этих классов: Кроме углеродной части в нефти имеются асфальто-смолистая составляющая, порфирины, сера и зольная часть. ... по которым в настоящее время передаются огромные количества природного, газа. Именно поэтому газ становится постепенно ... нефтях колеблется в широких пределах, но в среднем может быть принято на уровне 10—12%, тогда как содержание водорода в бензоле 7,7%. А что говорить о ...
При нахождении в воздухе в сравнительно больших количествах пары легкокипящих меркаптанов вызывают явление резкой интоксикации белых мышей. Токсические свойства уменьшаются по мере увеличения молекулярной массы меркаптанов. Наиболее ядовиты, исходя из этого, метил- и этилмеркаптаны. При содержании в воздухе этилмеркаптана в количестве 5 мг/л мыши умирали в течение несколько часов, при концентрации 1 мг/л наблюдались патологические нарушения в организме. Предельно-допустимая концентрация меркаптанов в непромышленной зоне составляет 1мг/м 3 . Исследование токсических свойств сульфидов и дисульфидов показало, что они гораздо менее ядовиты, чем меркаптаны. Их токсические свойства убывают по мере увеличения молекулярной массы так же, как и у меркаптанов.
Необходимость очистки производимых на нефтеперерабатывающих заводах моторных топлив от сернистых соединений и одновременная защита окружающей среды от загрязнения образующимися при этом отходами является для настоящего времени достаточно актуальной проблемой. Особенно опасны в этом отношении сульфидно-щелочные отходы, образующиеся при щелочной очистке компонентов моторных топлив от меркаптанов — токсичных и коррозионно-активных сернистых соединений — концентрация которых в топливе для карбюраторных, реактивных и дизельных двигателей не должна превышать 0,001-0,01 мас.%. В свою очередь, содержание сульфидов в промышленных стоках в естественных водоемах ограничивается величиной 0,001 мг/л.
В Российской Федерации около 10 млн. тонн нефтепродуктов подвергаются очистке от меркаптанов, в результате чего образуются сотни тысяч тонн сернистых щелочных стоков, оказывающих крайне негативное воздействие на окружающую среду. В частности, изменяется состав микрофлоры, так как сульфиды и меркаптиды натрия обладают токсичными и мутагенными действиями в концентрации 10~5 — 10 мг/л; индуцируют мутацию ауксотрофности и дыхательной недостаточности.
В настоящее время известны 3 способа удаления меркаптанов из моторных топлив: а) удаление всех сернистых соединений из очищаемого сырья, б) удаление меркаптанов и сероводорода из сырья, в) превращение меркаптанов в менее токсичные и коррозионно-активные соединения без удаления из сырья. Для удаления всех сернистых соединений используется главным образом гидроочистка, хотя есть способы, основанные на окислении. Недостатком этого способа является большая энергоемкость. Удаление сероводорода и меркаптанов, а также конверсия их в менее агрессивные соединения производится путем применения щелочных растворов. Наиболее прогрессивные методы удаления меркаптанов включают в себя процесс регенерации щелочи, вследствие чего расход ее снижается, но не устраняется. Наиболее известный способ борьбы с данными отходами «MEROX» включает в себя процесс каталитической окислительной регенерации щелочи, позволяющий резко сократить количество стоков, но не исключить их.
Полностью избавиться от сульфидно-щелочных отходов позволяет использование для демеркаптанизации бифункциональных катализаторов. Однако, существенным недостатком этого способа является низкая стабильность работы катализаторов вследствие быстрого снижения их активности. Это снижение активности может быть компенсировано повышением температуры, однако при этом снижается селективность процесса и резко увеличивается расход энергии для нагрева и охлаждения очищаемого сырья. Решение задачи активации катализатора без повышения температуры является необходимым условием решения актуальной задачи исключения образования и сброса сульфидно-щелочных стоков в окружающую среду.
Гетероатомные соединения нефти
... В группу гетероатомных соединений нефти включают смолисто-асфальтеновые вещества, содержащие в себе все гетероатомы нефти: кислород, азот ... щелочных металлов — это деэмульгаторы нефти и используются для ее обезвоживания. ^ Серосодержащие соединения Содержание сернистых соединений в нефтях ... подготовки, содержанию сероводорода и легких меркаптанов нефти подразделяют на классы типы, группы ...
1. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
Традиционный метод каталитического обессеривания — гидрообессеривание (HDS) с использованием катализаторов, таких как CoMo или NiMo, требует не только высоких температур, но и высокого давления. Также гидрообессеривание эффективно только на алифатических серных структурах, таких, как тиолы, тиоэфиры и дисульфиды и т.д. Серосодержащие ароматические соединения (в том числе тиофен и его производные) едва ли можно удалить этим процессом. По этой причине были разработаны альтернативные технологии сероочистки, в том числе экстракция, адсорбция и селективное окисление. Однако, использование летучих органических соединений (ЛОС) в качестве экстрагента приводит к более высокой степени экологичности и безопасности.
1 ЭКСТРАКЦИЯ
Экстракция сероочистки считается одним из лучших методов, поскольку имеет простую технологию. Начало количественному описанию экстракции (с химических позиций) положили Кольтгоф и Сендел, которые вывели в 1941 году уравнение, характеризующее экстракцию хелатов. Ирвинг и Уильямс развили эту теорию. Последующие интенсивные исследования привели к выяснению химизма большинства экстракционных процессов. Современные экстракционные методы достаточно универсальны. Трудно найти типы соединений, которые нельзя было бы экстрагировать. С помощью экстракции можно разделять многокомпонентные системы, причем эффективнее и быстрее, чем это достигается другими методами. Экстракционные методы пригодны для абсолютного и относительного концентрирования, извлечения в экстракт микроэлементов или матрицы, индивидуального и группового выделения элементов.
Экстракция — это процесс распределения вещества между двумя несмешивающимися растворителями. Одним из них обычно является вода, вторым — органический растворитель. Будучи гетерогенным процессом, экстракция подчиняется правилу фаз Гиббса:
+ F = K + 2,
где N — число фаз, F — число степеней свободы, K — число компонентов. При экстракции обычно две фазы (N = 2), одно распределяемое вещество (K = 1).
Следовательно, при постоянных температуре и давлении система моновариантна (F = 1).
Таким образом, если концентрация растворенного вещества в одной фазе постоянна, то его концентрация в другой фазе также постоянна. Соотношение между концентрациями растворенного вещества в каждой из фаз привело к формированию закона распределения.
Выполнение экстракционного разделения и концентрирования обычно не требует сложного и дорогостоящего оборудования. В лаборатории это чаще всего делительная воронка. С помощью воронки проводят так называемую периодическую экстракцию. Обычно водный раствор пробы и органический растворитель тщательно перемешивают встряхиванием вручную или с помощью механического устройства. После разделения фаз нижнюю фазу сливают через кран. Сильное встряхивание нежелательно, так как оно может привести к образованию эмульсий, что затрудняет разделение двух фаз. Если увеличение нужного компонента неполное, экстракцию повторяют, разделив фазы и прибавив к водной фазе новую порцию органического растворителя.
Понятие экстракции
... водной фазы в органическую. Для этого необходимо подобрать условия образования подходящих соединений (например, комплексов металлов), в виде которых компонент может находиться в органической фазе . 2. УСЛОВИЯ ЭКСТРАКЦИИ ВЕЩЕСТВА ... двух фаз. Если увеличение нужного компонента неполное, экстракцию повторяют, разделив фазы и прибавив к водной фазе новую порцию органического растворителя. Экстракция - ...
Экстракция — сложный физико-химический процесс. Теория экстракции находится на стыке различных разделов химии: химической термодинамики, теории растворов, химической кинетики, органической химии и координационной химии. Для описания экстракционных процессов необходимо также использовать теорию массопереноса. Задача экстракции состоит в том, чтобы полно и селективно перевести компонент из водной фазы в органическую. Для этого необходимо подобрать условия образования подходящих соединений (например, комплексов металлов), в виде которых компонент может находиться в органической фазе.
Условия экстракции вещества.
- Чтобы ион металла и другие заряженные частицы перешли в органическую фазу, необходимо нейтрализовать заряд. Ионы металла можно связать в незаряженный комплекс;
- комплексы, имеющие заряд, можно экстрагировать в виде ионных ассоциатов.
- Экстракция возможна, если растворимость экстрагирующегося соединения в органическом растворителе выше, чем в воде;
- чем больше энергия сольватации и меньше энергия гидратации, тем выше степень извлечения.
— Для того чтобы соединение было хорошо растворимо в органическом растворителе, необходимо обеспечить его гидрофобность, т.е. должны, как правило, отсутствовать гидрофильные группы (-SO 3 H, -COOH, -OH и др.) и внешняя органическая часть хелата должна быть достаточно объемистой и могла блокировать гидрофильную часть молекулы.
- С увеличением размера молекул экстрагирующегося соединения степень извлечения обычно повышается, поскольку крупные молекулы сильнее нарушают структуру воды.
- Экстракции способствует «сольватация» молекулами экстрагента. Например, экстракция ионов кадмия, кобальта и других двухзарядных ионов — оксихинолином в хлороформе обеспечивается образованием сольватов состава Ме(Ох) 2 ·nНОх.
- При экстракции ионных ассоциатов важны заряд и размер ионов;
- экстракция ухудшается с увеличением заряда и уменьшением размера ионов. При прочих равных условиях обычно лучше экстрагируются однозарядные ионы, хуже — двух- и особенно трехзарядные. В равных условиях более устойчивые комплексы экстрагируются лучше.
Способы осуществления экстракции.
Периодическая экстракция представляет собой экстракцию вещества из водной фазы отдельными порциями свежего экстрагента. При достаточно высоких значениях коэффициента распределения однократная экстракция позволят количественно извлечь вещество в органическую фазу. Эффективность однократной экстракции можно характеризовать степенью извлечения. Если однократная экстракция не обеспечивает достаточной степени извлечения, то Е можно повысить за счёт увеличения V(O) или прибегая к многократной экстракции. Часть вещества, оставшаяся в водной фазе после первой экстракции, будет равна

После любой n-кратной экстракции оставшуюся часть вещества в водной фазе можно рассчитать по формуле:

Флюидная экстракция комплексов урана из техногенных месторождений
... количество техногенных отходов практически не уменьшается. Глава 2. УСТАНОВКА ДЛЯ СВЕРХКРИТИЧЕСКОЙ ФЛЮИДНОЙ ЭКСТРАКЦИИ КОМПЛЕКСОВ УРАНА ИЗ ТЕХНОГЕННЫХ МЕСТОРОЖДЕНИЙ Украина обеспечена собственными урановыми ресурсами ... чёрной металлургии. 2. Месторождения наливные, образующиеся при заполнении впадин земной поверхности. Представителями этого типа ТМ являются: отходы обогащения руд (шламо- и ...
Отсюда степень извлечения после n-кратной
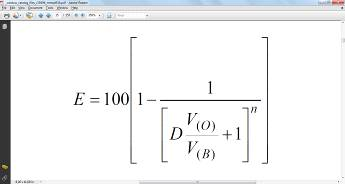
Вычисление числа ступеней экстракции для достижения заданной конечной концентрации экстрагируемого вещества осуществляют по формуле:
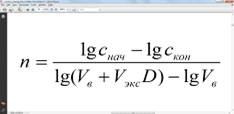
Расчет распределения вещества в зависимости от числа экстракций показывает, что для достижения максимальной степени экстракции число последовательных экстракций может быть не более 5-6 при D=1.
Периодическую экстракцию преимущественно проводят в делительной воронке. В делительную воронку вводят водный раствор, содержащий экстрагируемое соединение, и органический растворитель, не смешивающийся с водной фазой. Затем воронку энергично встряхивают для обеспечивания хорошего контакта фаз. После встряхивания фазы разделяют.
Непрерывная экстракция — осуществляется при непрерывном и относительном перемещении двух фаз; одна из фаз, обычно водная, остаётся неподвижной.
Противоточная экстракция. Последовательность операций в противоточном распределении заключается в том, что верхняя, как правило, органическая фаза переносится последовательно через серию экстракционных трубок и в каждой из них контактирует со свежими порциями нижней водной фазы до установления равновесия. Таким образом, на каждой стадии происходит распределение веществ между свежими порциями обеих фаз. Процесс установления равновесия и переноса повторяют n раз. Противоточную экстракцию применяют для разделения сложных смесей, а также для выделения малых количеств компонентов из больших объёмов исходного материала. Например, с помощью прибора, состоящего из 40 трубок, был выделен протоген из 4 т говяжьей и свиной печени. Есть и другие способы осуществления противоточной экстракции.
Экстракционные процессы по типу используемого экстрагента можно разделить на три группы:
- экстракция кислотными (катионообменными) экстрагентами. К ним относятся карбоновые и нафтеновые кислоты;
- фосфорорганические и сульфокислоты;
- хелатообразующие экстрагенты: β-дикетоны, купфероны, гидроксамовые кислоты, 8-оксихинолин, диметилглиоксим, дифенилтиокар- базон, диэтилдитиокарбаминаты;
- основными (анионообменными) экстрагентами. Это соли третичных аминов и четвертичных аммониевых оснований (R 4 N+X-);
- соли тетрафенил- фосфония ((C6 H5 )4 P+X-) и тетрафениларсония ((C6 H5 )4 As+X-);
- нейтральными экстрагентами. К ним относятся эфиры: диэтиловый, 2,2-дихлордиэтиловый;
- кетоны: метилизобутилкетон, циклогексанон;
- диантипирилметан и его аналоги;
- трибутилфосфат;
- сульфиды (RR/S);
- сульфоксиды (RR/SО);
- фосфаты ((RO) 3 PO);
- фосфонаты ((RO)2 RPO);
- фосфинаты ((RO)R2 PO);
- фосфиноксиды (R3 PO);
- фосфины ((С6 H5 )3 P) и др.
2 АДСОРБЦИЯ
Адсорбция (от лат. ad — на, при и sorbeo — поглощаю) — преимущественное концентрирование молекул газа или растворённого в жидкости вещества (адсорбата) на поверхности жидкости или твёрдого тела (адсорбента), а также растворённого в жидкости вещества на границе её раздела с газовой фазой. Является частным случаем сорбции и одним из важнейших типов поверхностных явлений.
Производство серной кислоты из серы
... производства целевого продукта и экологической безопасности производства 3.1 Общая схема сернокислотного производства Производство серной кислоты из серосодержащего сырья включает несколько химических процессов, в которых происходит изменение степени окисления ... и др.), сероводород и ряд других сернистых соединений. Традиционно основные источники сырья - сера и железный (серный) колчедан. Постепенно ...
Явление адсорбции связано с тем, что силы межмолекулярного взаимодействия на границе раздела фаз не скомпенсированы, и, следовательно, пограничный слой обладает избытком энергии — свободной поверхностной энергией. В результате притяжения поверхностью раздела фаз находящихся вблизи неё молекул адсорбата свободная поверхностная энергия уменьшается, т. е. процессы адсорбции энергетически выгодны.
В зависимости от характера взаимодействия молекул адсорбата и адсорбента различают физическую адсорбцию и хемосорбцию. Физическая адсорбция обусловлена силами межмолекулярного взаимодействия и не сопровождается существенным изменением электронной структуры молекул адсорбата. Физическая адсорбция может быть как монослойной (с образованием мономолекулярного слоя), так и полимолекулярной (многослойной).
При А. электролитов из их растворов обычно возникает двойной электрический слой. Если жидкий адсорбат смачивает пористый адсорбент, то в порах последнего может происходить капиллярная конденсация. При физической адсорбции адсорбирующие молекулы обычно обладают поверхностной подвижностью.
При хемосорбции между атомами (молекулами) адсорбента и адсорбата образуется хим. Связь, таким образом, хемосорбцию можно рассматривать как химическую реакцию, область протекания которой ограничена поверхностным слоем. В некоторых случаях на одной поверхности могут протекать оба типа адсорбции одновременно. В случае не слишком пористых адсорбентов физическая адсорбция имеет место, как правило, при температуpax ниже критической температуры конденсации адсорбата, хемосорбция же чаще всего протекает при гораздо более высоких температурах. Однако в некоторых системах физическая адсорбция может протекать при температуpax, значительно превышающих критич. температуру конденсации адсорбата. Как и любые химические реакции, процессы хемосорбции носят специфичный характер (т. е. адсорбент хемосорбирует не любые молекулы, а лишь те, которые вступают в реакцию с атомами поверхности); в некоторых случаях специфичность может проявляться и при физической адсорбции.
Количественной характеристикой адсорбции является величина Г, представляющая собой избыток адсорбата, приходящийся на единицу площади поверхностного слоя, по сравнению с количеством адсорбата в единицу объёма фазы адсорбента. Отношение
меркаптан топливо адсорбция окислительный
называется степенью (или долей) покрытия поверхности ( -предельно возможная величина монослойной адсорбции для данной системы).
Процессы адсорбции почти всегда сопровождаются выделением теплоты, называемой теплотой адсорбции, которая возрастает с увеличением прочности связи адсорбат — адсорбент и составляет обычно 8-25 кДж/моль (иногда до 80 кДж/моль) для физической и, как правило, превышает 80 кДж/моль при хемосорбции. Если хемосорбция сопровождается диссоциацией адсорбированных молекул, может наблюдаться поглощение тепла. По мере заполнения поверхности теплота адсорбции обычно уменьшается в результате неоднородного распределения свободной энергии на поверхности или латерального взаимодействия молекул в адсорбированном слое. Для адсорбентов, обладающих несколькими типами адсорбирующих центров, теплота адсорбции может быть различной для разных типов центров, и распределение свободной энергии на поверхности является дискретно-неоднородным. При переходе к полимолекулярной адсорбции теплота адсорбции понижается до величины, близкой к теплоте конденсации адсорбата. Если теплота адсорбции сравнима с поверхностной энергией адсорбента, то в процессе адсорбции может существенно меняться кристаллическая структура поверхности твёрдого тела, причём при физической адсорбции перестройке подвергаются в основном поверхности молекулярных кристаллов, а в случае хемосорбции изменение поверхностной структуры наблюдается даже для металлов и ионных кристаллов.
Приоритеты в качестве дизельного топлива
... солярка происходит из немецкого Solarцl <#"justify">Химический состав дизельного топлива определяется происхождением нефти и технологией получения топлива. Дизельное топливо является сложной смесью парафиновых (10-40%), нафтеновых ... применяют специальный орт топлива с температурой вспышки 65-80 градусов. 3.4 Содержание серы и воды Сернистые соединения, непредельные углеводороды и металлы ...
Обратный адсорбции процесс, при котором адсорбированные частицы покидают поверхность адсорбента, называют десорбцией. Десорбция происходит в результате колебательного движения адсорбированных молекул вдоль направления действия силы притяжения между адсорбатом и адсорбентом. Период таких колебаний t0 обычно составляет 10-13 с. Скорость адсорбции и скорость десорбции могут быть рассчитаны методами статистической термодинамики. Скорость медленных процессов хемосорбции в большинстве случаев описывается уравнением
где — кол-во адсорбированного вещества, а и — константы, зависящие от температуры. При равенстве скоростей адсорбции и десорбции устанавливается адсорбционное равновесие. Средняя продолжительность времени, которое частица находится в адсорбированном состоянии в равновесных условиях (время адсорбции),
где Q — теплота адсорбции, R — универсальная газовая постоянная, Т — абсолютная температура. Принято считать, что адсорбция имеет место в том случае, когда достигает величины нескольких периодов колебаний адсорбированной молекулы — время, за которое между ней и поверхностью успевает установиться энергетическое равновесие. Обычно время физической адсорбции составляет 10-12-10-6 с, а время хемосорбции — свыше 102 с. Время адсорбции служит критерием обратимости процесса адсорбции.
Эффективность окислительного обессеривания углеводородного топлива существенно повышается, когда за процессом окисления следует адсорбция продуктов окисления на твердом адсорбенте. Так, гетерогенная система на основе модифицированного алюмосиликата может быть использована в качестве адсорбента для обессеривания и деазотирования модельного топлива, составленного из н-тетрадекана и ксилолов, и содержащего дибензотиофен, анилин, индол и карбазол. Адсорбция N- и S-содержащих соединений на поверхности адсорбента происходит с образованием комплексов с переносом заряда и подавляется присутствием в топливе ароматических углеводородов. Максимальная степень удаления азот- и сероcодержащих соединений 62,5% достигается сочетанием окисления с процессом адсорбции продуктов окисления на поверхности адсорбента. В исследовательской работе [6] использование силикагеля для адсорбции продуктов окисления прямогонной дизельной фракции и газойля каталитического и термического крекинга пероксидом водорода в присутствии смеси серной и уксусной кислот позволило снизить содержание серы в топливе с 482 ppm до 50 ppm. Адсорбционное обессеривание модельных дизельных топлив с содержанием органической серы 500 мг/г в виде тиофена, тетрагидротиофена и 4,6-диметилбензотиофена было осуществлено в статической системе и в проточном реакторе на цеолите, обработанном раствором гексафторогаллатом аммония и содержащем ионы галлия. По данным авторов этой работы арены и олефины тормозят адсорбцию сернистых соединений, величина которой для тиофена меньше, чем для тетрагидротиофена и 4,6-диметилдибензотиофена.
Окислительное обессеривание легкого газойля с содержанием серы 39 ppm в присутствии гетерогенного катализатора 16% MoO 3 /Al2 O3 было осуществлено действием трет-бутилгидропероксида [7].
Окислительная активность сернистых соединений в топливе возрастает, когда соотношение О/S увеличивается до 15, при дальнейшем росте соотношения она слабо снижается. Для каждого индивидуального соединения — дибензотиофена, 4,6-диметилдибензотиофена и С3-дибензотиофена окисление представляет собой реакцию первого порядка с энергией активации ~32 кДж/моль. Последующая адсорбционная обработка обессеренного окислением топлива снижает содержание серы до 5 ppm и ниже. Деазотирование с помощью этой методики позволяет снизить содержание азота в топливе с 13,5 до 0,8 ppm.
Сочетание пероксидного окисления сернистых соединений в моторном топливе в присутствии гетерогенных носителей с последующим адсорбционным удалением продуктов окисления описано также в ряде патентов [8,9]. В качестве гетерогенных катализаторов использовали твердые кислотные катализаторы и/или активированный уголь, катализаторы содержат оксид переходного металла. Кислотный катализатор выбирают из следующей группы соединений: сульфат циркония, сульфат алюминия, сульфатированный оксид олова, оксид железа, молибдат циркония и молибдат оксида олова. Метод адсорбционного обессеривания на адсорбенте Ni/SiO 2 -Al2 O3 был использован для удаления серы из реактивных топлив с исходным содержанием серы 736 и 380 ppm. Предварительное удаление из топлива следов 2,3,7-триметилбензотиофена улучшило адсорбционную способность адсорбента в 2,5 раза. Авторы работы достигли оптимальной адсорбционной емкости адсорбента в 10 мг S/г при размере его частиц 0,15-0,25 мм без создания дополнительного давления при элюировании растворителя через слой адсорбента.
Эффективность окислительного обессеривания, представляющего собой сочетание каталитического окисления сернистых соединений молекулярным кислородом в присутствии гетерогенного катализатора и адсорбции на активированном угле в мягких условиях (25 °С) была также продемонстрирована в работе [10].
Более активными, по данным авторов, являются бензотиофены с объемными алкильными заместителями; преимуществом такой системы является отсутствие необходимости проводить окисление в двухфазной системе вода-моторное топливо. Для глубокого обессеривания дизельного топлива с содержанием серы 1786 ppm при 373 K был использован алюмосиликатный адсорбент MCM-41 с различным соотношением SiO 2 /Al2 O3 (100, 50 и 30).
Адсорбционная способность сернистых соединений уменьшается с возрастанием мольного соотношения SiO2 /Al2 O3 в адсорбенте, а эффективность удаления серы составляет 95% на адсорбенте с мольным соотношением SiO2 /Al2 O3 = 50 на начальной стадии и дополнительно 75% после совокупного объема истечения элюента 17 мл. Повышение температуры является негативным фактором, а присутствие ионов Сu+ положительно воздействует на процесс адсорбции вследствие образования π-комплексов между сернистыми соединениями и ионами меди. Пероксидное окисление модельных сернистых соединений: тиофена, 2-метилтиофена, бензотиофена, дибензотиофена, 4-метилдибензотиофена и 4,6-диметилдибензотиофена и окислительное обессеривание дизельного топлива были проведены действием трет-бутилгидропероксида в присутствии гетерогенного катализатора MoO3 /Al2 O3 в статическом реакторе. Этот катализатор обладает высокой активностью, но постепенно дезактивируется из-за выщелачивания металла и адсорбции сульфонов. Аналогичная методика была использована для обессеривания сырой нефти, она основана на применении смеси алкилгидропероксида, кислоты и порошкообразного оксида железа. В ходе процесса образуется пероксокислота и гидроксильные радикалы, присутствие которых обусловлено наличием оксогидроксида железа, обладающего, по мнению авторов работы, особым сродством к углеводородной среде.
Значительное распространение для удаления сернистых соединений из моторных топлив получили гетерогенные системы, состоящие из разнообразных твердых носителей (соли, оксиды, активированный уголь, цеолиты) и пероксидных окислителей (пероксид водорода или алкилгидропероксиды).
Так, в работе [11] изучалось окисление пероксидом водорода на ванадиевом катализаторе модельных сернистых соединений: 2-метилтиофена, 2,5-диметилтиофена, бензотиофена, дибензотиофена, 4-метилдибензотиофена и 4,6-диметилдибензотиофена. На примере модельного дизельного топлива, приготовленного из гексадекана и указанных сернистых соединений, было установлено уменьшение окислительной активности сернистых соединений в следующей последовательности:
- метилдибензотиофен >
- 2-метилтиофен >
- 2,5-диметил-тиофен >
- 4,6-диметилдибензотиофен.
Избыток окислителя вызывает повышенное образование воды, что, в свою очередь, тормозит окислительное обессеривание. Авторы справедливо делают вывод о необходимости ограничения количества добавляемого в реакционную смесь пероксида водорода. Трет-Бутилгидропероксид использовался в качестве селективного окислителя различных сераорганических соединений (метилфенилсульфид, дифенилсульфид, 4-метилдибензотиофен, 2,5-диметилтиофен) на гетерогенных катализаторах CoAPO-5 (APO-алюминофосфат, Сu/Al=0,15) и 15% MoO 3 /Al2 O3 . Cкорость окисления сернистого соединения растет с увеличением электронной плотности на атоме серы и существенно выше на кобальтовом, чем на молибденовом катализаторе. При высоких степенях превращения сернистых соединений сульфоны являются единственными продуктами окисления, тогда как при низких образуются также небольшие количества сульфоксидов. По данным спектроскопии в УФ и видимой областях частицы, участвующие в окислительно-восстановительной реакции, контролируют также лимитирующую стадию всего процесса. Из других твердых носителей, использованных для окисления тиофеновых соединений пероксидом водорода, можно отметить силикат титана (TS-1) [12].
На ход окисления тиофена при 60 °С влияет наличие кристаллической структуры TS-1 и обработка носителя HCl. Оксиды марганца и кобальта, нанесенные на Al 2 O3 , также катализируют окисление воздухом сернистых соединений, присутствующих в дизельном топливе, при 130-200 °С и атмосферном давлении. При последующей экстракции продуктов окисления полярным растворителем содержание серы в дизельном топливе снижается до 40-60 ppm. Сравнение с данными по гидрообессериванию показывает, что наиболее активные сернистые соединения в этом процессе также проявляют высокую активность и в окислительном обессеривании.
Высокая степень удаления серы (98%) была достигнута в мягких условиях действием пероксида водорода в присутствии полимолибдатов, нанесенных на Al 2 O3 [13].
Контроль за приготовлением катализатора методами ИК, КР, РСА, 31P и 27Al ЯМР показал, что в процессе синтеза происходит разложение катализатора, на его поверхности образуются гепта- и октамолибдаты, а также фосфат-ионы. Эффективным катализатором окислительного обессеривания дизельного топлива газолина является 0,06% Ag/TS-1, приготовленный методом пропитки. В его присутствии при действии водного раствора пероксида водорода содержание серы в топливе снизилось с 136,5 мг/л до 18,8 мг/л после 4-часовой обработки.
Из титансодержащих катализаторов, активных в окислительном обессеривании дизельного топлива пероксидом водорода, можно также отметить предложенный китайскими авторами Ti/HMS, который проявляет высокую активность в окислении диметилбензотиофена [14].
Степень удаления дибензотиофена из модельной смеси с н-октаном составила 90%, а содержание серы в дизельном топливе было снижено при 333 К с 226 до 23 ppm при мольном соотношении H 2 O2 /S =4:1.
Молибденсодержащие катализаторы являются одними их самых эффективных гетерогенных систем для окислительного обессеривания дизельного топлива. Ряд таких каталитических систем был предложен недавно мексиканскими учеными [15], которые исследовали окислительное обессеривание действием пероксида водорода в присутствии Mo/γ-Al 2 O3 . Активность катализатора в удалении серы зависит главным образом от присутствия гепта- и октамолибдатов на его поверхности и от добавления фосфатов. Использование такого катализатора при 333 K позволяет снизить содержание серы в топливе с 320 до 10 ppm. Одной из ключевых стадий предлагаемого авторами механизма окислительного обессеривания является образование гидропероксимолибдатных фрагментов на поверхности катализатора с последующим их взаимодействием с производными дибензотиофена и окислением последних до сульфонов.
Распространенным носителем для гетерогенного окислительного обессеривания является активированный уголь [16,17]. Его применение позволяет удалить из газообразного топлива сероводород при 150 о С окислением кислородом воздуха, а также осуществить окислительное обессеривание дизельного топлива действием пероксида водорода. Чем выше адсорбционная способность активированного угля, тем выше его активность в окислении дибензотиофена, добавление муравьиной кислоты ускоряет окисление. Использование такой системы позволяет понизить содержание серы в дизельном топливе с 800 до 142 ppm. Дополнительная адсорбционная обработка топлива активированным углем понижает содержание в нем серы до 16 ppm.
Активированный уголь для окислительного обессеривания дизельного топлива пероксидом водорода был также использован в работе [18], где авторам удалось удалить 98% серы с потерями, не превышающими 3,5%. Эффективными в окислении дибензотиофенов и окислительном обессеривании кислородом воздуха дизельного топлива являются системы из ионов металла и альдегидов [19,20]. Ацетаты кобальта(II), никеля(II) или марганца(II) в присутствии н-октаналя и н-гексаналя обеспечивают 99% конверсию дибензотиофена в сульфон за 15 мин при 40 °С. Сочетание окисления с адсорбцией на Al 2 O3 или SiO2 и экстракцией полярным растворителем позволяет достичь степени удаления серы из топлива более 97%. Приготовленные из ацетатов Co и Mn нанесенные катализаторы Co/Al2 O3 и Mn/Al2 O3 в присутствии альдегидов обеспечивают достаточно высокую активность в окислении дибензотиофена (до 62%) системой кислород/альдегид; в то же время никелевый катализатор Ni/Al2 O3 , также приготовленный из ацетата никеля, не проявил активности в этой реакции.
Использование в работе [21] различных твердых катализаторов для окислительного обессеривания дизельного топлива пероксидом водорода позволило установить следующий ряд их активности:
- >
- Cr 2 O3 >
- MnxOy >
- Co — Mo/Al2 O3
Различные оксиды марганца, Mn 3 O4 , Mn2 O3 и MnO2 , имеют примерно равную активность, независимо от концентрации пероксида водорода. Авторы полагают, что каталитическое разложение Н2 О2 конкурирует с окислительным обессериванием, но все же за короткое время (до 10 мин) можно достигнуть 60-70%-й конверсии тиофена. Высокую каталитическую активность в окислении сульфидов до сульфоксидов трет-бутилгидропероксидом проявляет ацетилацетонатный комплекс ванадия VO(acac)2 , нанесенный на TiO2 . По данным этих же авторов, использование по отдельности TiO2 и VO(acac)2 позволяет достичь той же степени конверсии сульфида (99%), но с большим (до 44%) содержанием сульфона в продуктах окисления.
Гетерогенная система на основе молекулярных сит SBA-15, содержащая фосфоромолибденовую кислоту H 3 PW12 O40 , была использована в окислительном обессеривании трет-бутилгидропероксидом модельного топлива, составленного из изооктана и бензотиофена [22].
Эта система, действуя как катализатор и адсорбент одновременно, может удалять из топлива до 90% дибензотиофена в одну стадию. Изучение катализатора методом ИК спектроскопии показало, что на его поверхности в ходе десульфуризации возникают интермедиаты с пероксидными фрагментами [O-О]. Хорошие результаты в окислении производных бензотиофена показал мезопористый титансиликатный катализатор TS-1, представляющий собой упакованные нанокристаллы [23].
Катализатор получали методом нанокастинга нанопористого углерода CМК-3. В сравнении с обычным мезопористым титансиликатом TS-1 нанокристаллический катализатор проявляет более высокую активность в окислении тиофена и диметилбензотиофенов пероксидом водорода.
Для окислительного обессеривания синтетического дизельного топлива действием H 2 O2 при атмосферном давлении и 60 °С пригодны нанесенные ванадиевые катализаторы V2 O5 /Al2 O3 и V2 O5 /TiO2 [24].
Присутствие в топливе азотсодержащих соединений препятствует адсорбции на поверхности катализаторов дибензотиофена, при этом активность катализатора в зависимости от типа содержащихся в топливе азотистых соединений уменьшается в следующей последовательности: хинолин > индол > карбазол.
4 ОБЕССЕРИВАНИЕ ИОННЫМИ ЖИДКОСТЯМИ
Ионные жидкости (ILS) широко используются в зеленой химии. Ионные жидкости имеют следующие характеристики: нелетучесть, невоспламеняемость и высокая термическая стабильность. В результате, ионные жидкости считаются зелеными растворителями и используются в процессах катализа, химического синтеза и разделения. Об использовании ионных жидкостей в качестве экстрагентов для удаления ароматических соединений серы в добывающей сероочистки было изложено в последних исследованиях. Bösmann впервые описал, что ионная жидкость имидазолия может быть использована как экстрагент. Большинство литературы описывает ионные жидкости просто как экстрагенты; результаты показывают, что ионные жидкости могут действенно экстрагировать серу от масляной фазы. Wen Hen Lo и его коллеги скомбинировали методы окисления и добычи, используя раствор Н 2 О2 — уксусная кислота в качестве окислителя, а [BMIM] BF4 и [BMIM] PF6 — как экстрагент; этот процесс увеличивает скорость сероочистки примерно на порядок по сравнению с показателями извлечения с RTILs.
Согласно докладам, наличие кислоты может привести к увеличению сероочистки, но когда дело доходит до промышленного применения, оборудование будет разъедается кислотой. В работе учёных использовался тип пиридиния [BPy] BF 4 как экстрагент, который смешивается с перекисью водорода в качестве окислителя на первое время. Изучалось обессеривание тиофена и дибензотиофена (DBT) в модели нефти, а также кипение каталитического крекинга (FCC) бензина.
Особенно эффективны ионные жидкости, которые содержат ионы Cu(I) и Ag(I), ввиду их значительной склонности к образованию π-комплексов с тиофеновыми производными [25].
Для обессеривания модельного топлива использовали ионные жидкости, полученные взаимодействием 1-бутил-3-метилимидазолийхлорида с безводным порошкообразным CuCl, содержащие в качестве анионов частицы CuCl 2 — , Сu2 Cl3 — и Cu3 Cl4 — , устойчивые к действию влаги и стабильные на воздухе. Эти системы показали высокую обессеривающую способность при очистке бензина. Например, после его экстракции ионной жидкостью BMImCu2 Cl3 cтепень удаления сернистых соединений достигала 23%, тогда как с помощью ионной жидкости состава BMImBF4 эта же величина не превышала 11%. Соединения с высокой комплексообразующей способностью, растворенные в бензине, тормозят экстракцию сернистых соединений ионными жидкостями и снижают степень удаления серы.
Как правило, сами ионные жидкости без окислителя не позволяют достичь высокой степени удаления серы. Например, пероксовольфрамовые и пероксомолибденовые комплексы [WO(O 2 )2 Phen H2 O] и [MoO(O2 )2 Phen], (где Рhen — 1,10-фенантролин), иммобилизованные в ионные жидкости (1-метил-3-бутилимидазолий гексафторфосфат, 1-бутил-3-метилимидазолий гексафторфосфат, 1-н-октил-3-метилимидазолий гексафторфосфат и тетрафторборат), только экстрагируют дибензотиофен, но не активны в его окислении [26].
Добавление 30% Н2О2 в ионную жидкость создает условия для каталитического окисления и экстракции, и степень удаления общей серы повышается до 99%. В отсутствие ионной жидкости такие фенантролиновые комплексы не позволяют достичь степени удаления серы выше 50%, что указывает на преимущества метода, сочетающего каталитическое окисление и экстракцию. Исследование экстракционной способности N-метил-N-метилимидазолий диметилфосфата [MMIM][DMP] и N-бутил-N-метилимидазолий дибутилфосфата [BMIM][DBP] в широком диапазоне концентраций серы в дизельном топливе показало, что растворимость дибензотиофена и бензотиофена в водных растворах ионных жидкостей при 25 °С меняется в следующем порядке:
- [BMIM][DBP] >
- [EMIM][DEP] >
- [MMIM][DMP].
При этом дибензотиофен растворяется лучше бензотиофена [27].
Из испытанных ионных жидкостей наиболее подходит для обессеривания топлива [EMIM][DEP], которая обладает относительно высокой способностью к удалению серы, низкой растворимостью в топливе и мало влияет на другие свойства топлива. Интересным представляется использование для обессеривания бензинов ионных жидкостей, синтезированных из органических кислот (муравьиная, уксусная и бензойная) и азотистых оснований (анилина, пиперидина и диэтиламина) [28].
После трехкратной экстракции бензина каталитического крекинга указанными ионными жидкостями содержание серы в бензине было снижено с 240 до 30 ppm, а содержание ароматических углеводородов с 26 до 14%. Ионные жидкости могут повторно использоваться для обессеривания после регенерации их из экстракта обработкой избытком низкокипящих парафинов.
Высокая эффективность ионных жидкостей для обессеривания дизельного топлива была продемонстрирована на примере использования ионных жидкостей, содержащих в качестве катиона 1-бутилметилимидазолий, а в качестве анионов — тетрафторборат, гексафторфосфат, октилсульфат, этилсульфат и диметилфосфат [29].
Предложенная технологическая схема, содержащая ступени экстракции и регенерации ионной жидкости, обеспечивает снижение содержания серы в дизельном топливе с 500 до 10 ppm.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез ионной жидкости. Ионная жидкость тетрафторборат N-бутилпиридиния [BPy] BF 4 была подготовлена с использованием методов, описанных в литературе. В общем случае, пиридин (0,2 моль), бутилбромид (0,2 моль) и циклогексан (50 мл) перемешивали в круглодонной колбе при 60°C в течение 12 ч. Белый осадок (N-бутилпиридиний бромид) отфильтровывали и выпаривали в вакуумной сушильной печи. Ионную жидкость получали путем смешивания N-бутилпиридиний бромида (0,2 моль) и тетрафторбората натрия (0,2 моль) с ацетоном (100 мл) в круглодонной колбе и в результате перемешивания смеси при комнатной температуре в течение 12 ч. Осадок отфильтровывали, а затем растворитель удаляли, в роторном испарителе оставалась желаемая желтая ионная жидкость.
Синтез бутилбромида: В круглодонную колбу емкостью 50 мл помещали раствор 7 мл бутилового спирта в 6 мл воды, затем при постоянном перемешивании и охлаждении постепенно приливали 15 мл концентрированной серной кислоты (пл.=1,84).
После чего реакционную смесь охлаждали до комнатной температуры и к ней при перемешивании добавляли 12г тонкорастертого бромида калия. Колбу присоединяли к нисходящему холодильнику. Реакционную смесь нагревали на песчаной бане, пока в приемник с водой не перестали переходить маслянистые капли, опускающиеся на дно. Бромистый бутил отделяли от воды с помощью делительной воронки, сушили хлоридом кальция и перегоняли из колбы Вюрца, собирая в сухой приемник, помещенный в холодную воду. Выход 8,5 г t кип. 38-39 о С при 760 мм рт.ст.
+ H 2 SO4 → KHSO4 + HBr
C 4 H9 OH + HBr C4 H9 Br + H2 O
CH 3 -CH2 -CH2 -CH2 -OH + HO-SO2 -OH CH3 -CH2 -CH2 -CH2 -O-SO2 -OH + H2 O
CH 3 -CH2 -CH2 -CH2 -O-SO2 -OH + HBr → CH3 -CH2 -CH2 -CH2 — Br + H2 SO4
Результаты и обсуждения.
По данным сайта www.ahmadullins.com <http://www.ahmadullins.com/>, содержание общей и меркаптановой серы в нефтях и газоконденсатах астраханского месторождения таково:
[S] % мacc.1.38
[SRSH] % мacc.0.19
[SRSH] % отн.13.8СН з SH % мacc.0.0010С2 Н5 SH % мacc.0.0160
При определении содержания серы в различных моделях бензина методом энергодисперсионной рентгенофлуоресцентной спектрометрии (АСЭ — ГОСТ Р51947-2002) были получены следующие результаты:
Таблица 1.
|
Образец |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
% масс. |
0,0116 |
0,0139 |
0,0914 |
0,1026 |
0,0123 |
2,7302 |
1,8808 |
1,0015 |
1,0037 |
Окислительная сероочистка бензина с помощью ионной жидкости.
Оба катиона, ионной жидкости и тиофена (равно как и его производные) имеют ароматические кольца. Когда катион ионной жидкости, который имеет ароматическое структурное кольцо с большой π-связью, взаимодействует с серосоединением тиофеном, образуется комплекс и тиофен извлекается на фазe ионной жидкости из-за легко поляризованных молекул серы.
Система экстракции и система окислительной экстракции была применена к модельной системе октан-тиофен, содержащей 780 мкг / мл серы. Условия реакции для экстракционной сероочистки: соотношение масс ионной жидкости и модели составляет 1:1, реакции в течение 30 мин при 25 ° C.
Условиями реакции окислительной экстракции десульфоризации являются: соотношение масс ионной жидкости и модели бензина 1:1, соотношение Н 2 О2 и ионной жидкости 0,4:1, время и температура те же. Концентрации серы в модели бензина до и после реакции приведены в таблице 2, где видно, что содержание серы в модели бензина может быть уменьшено на 25,9% (экстракционная сероочистка) и 36,3% (окислительно-экстракционная сероочистка).
Бензин содержит много неароматических серных компонентов, таких, как тиоловые сульфиды, которые не могут быть извлечены эффективно с помощью ионной жидкости. С другой стороны, ароматические углеводороды и олефины в бензине также хорошо влияют на экстракцию.
Таблица 2: Результаты сероочистки бензина (модель октан + тиофен).
|
Система ионной жидкости |
Содержание серы |
Десульфоризация |
|
Экстракция |
780 мг/мл |
25,9% |
|
Окислительная |
780 мг/мл |
36,8% |
Условия: a: температура = 25 °C; V (модель) / V (ионная жидкость) = 1:1; время = 30 мин; b: температура = 25 °C; V (модель) / V (ионная жидкость) / V(H 2 O2 ) = 1:1:0.4; время = 30 мин.
ЗАКЛЮЧЕНИЕ
Ионная жидкость [BPy] BF 4 оказалась эффективной при извлечении компонентов серы из бензина при комнатной температуре. Сероочистка процесса была достигнута после добавления Н2 О2 к экстракционной системе; окисляемость Н2 О2 была использована для окисления компонентов серы до соответствующих сульфонов, которые были извлечены ионной жидкостью. Использованная ионная жидкость может быть регенерирована через реэкстракции в тетрахлорметане. Это доказывает возможности сероочистки как простого, легкого и экологически безопасного метода в промышленности будущего.
СПИСОК ЛИТЕРАТУРЫ
[Электронный ресурс]//URL: https://inzhpro.ru/kursovaya/demerkaptanizatsiya-nefti/
1. Золотов Ю.А., Кузьмин Н.М. Концентрирование микроэлементов. М.: Химия, 1982. 288 с.
- Золотов Ю.А. Экстракция в неорганическом анализе. М.: Изд-во МГУ, 1988. 82 с.
- Золотов Ю.А.
Экстракция внутрикомплексных соединений. М.: Наука, 1968. 313 с.
- Харитонов Ю.А. Комплексные соединения // Соросовский Образовательный Журнал. 1996. № 1. С. 48-56.
- Будников Г.К., Троепольская Т.В., Улахович Н.А.
Электрохимия хелатов металлов в неводных средах. М.: Наука, 1980. 192 с.
- Шарипов А.Х., Нигматуллин В.Р. Xимия и технол. топлив и масел , 2005, № 3, с. 42-44.
- Ishihara A., Wang D., Dumeignil F., Amano H., Weihua Qian E., Kabe T.
Appl. Catal. A: General, 2005, v. 279, № 1-2, p. 279-287.
- Патент США № 60226049, 2006.
- Патент ЕP № 1715025, 2006.
- Ma C., Zhou A., Song C.
Catal. Today, 2007, v. 123, № 1-4, p. 276-284.
- Caero L.C., Hernandez E., Pedraza F., Murrieta F. Catal. Today, 2005, v. 107-108, p. 564-569.
- Lingyan Kong, Gang Li, Xiangsheng Wang.
Catal. Today, 2004, v. 93-95, p. 341-345
- Garsia-Gutierrez J.L., Fuentes G.A., Hernandez-Teran M.E., Murrieta F., Navarrete J., Jimenez-Cruz F. Appl. Catal. A:General, 2006, v. 305, № 1, p. 15 20.
- Yun Wang, Gang Li, Xiangsheng Wang, Changzi Jin.
Ibid., 2007, v. 21, № 3, p. 1415-1419.
- Garsia-Gutierrez J.L., Fuentes G.A., Hernandez-Teran M.E., Garsia P., Murrieta-Guevara F., Jimenez-Cruz F. Appl. Catal. A: General, 2008, v. 334, p. 366-373.
- Xianxian Wu, Shwartz V., Overbury S.H., Armstrong T.R.
Energy and Fuels, 2005, v. 19, № 5, p. 1774-1782.
- Guoxian Yu, Shanxiang Lu, Hui Chen, Zhongman Zhu. Ibid., 2005, v. 19, № 2, p. 447-452.
- Guoxian Yu, Shanxiang Lu, Hui Chen, Zhongman Zhu.
Carbon, 2005, v. 43, № 11, p. 2285-2294.
- Murata S., Murata K., Kidena K., Nomura M. Energy and Fuels, 2004, v. 18, № 1, p. 116-121.
- Dumont V., Oliviero L., Mauge F., Houalla M.
Catal. Today, 2008, v. 130, № 1, p. 195-198.
- Zapata B., Pedraza F.,Valenzuella M.A. Ibid., 2005, v. 106, № 1-4, p. 219-221.
- Lina Yang, Jian Li, Xingdong Yuan, Jian Shen, Yutai Qi.
J. Mol.Catal. A: Chemical, 2007, v. 262, № 1-2, p. 114-118.
- Yunming Fang, Haoguan Hu. Catalysis Commun., 2007, v. 8, № 5, p. 817-820.
- Caero L.C., Jorge F., Navarro A., Gutierrez-Alejandre A.
Catal. Today, 2006, v. 116, № 4, p. 562-568.
- Hernandez A.J., Yang R.T. Ind. Eng. Chem. Res., 2003, v. 42 , p. 123-129.
- Zhu W., Li H., Jiang X., Yan Y.,Lu J., Xia J.
Ibid, 2007, v. 21, № 5, p. 2514-2516.
- Nie Y., Li C., Sun A., Meng H., Wang Z. Ibid., 2006, v. 20, № 5, p. 2083-2087.
- Азизов А.Г., Гусейнова А.Д., Ибрагимова М.Д., Азмамедов Н.Г., Гусейнова И.С., Эйвазов Э.З., Юнусов С.Г.
Нефтепереработка и нефтехимия, 2007, № 6, с. 25-26.
- Esser J., Wasserscheid P., Jess A. Green Chemistry, 2004, № 6, p. 316-322
- Dishun Zhao, Yanan Wang, Erhong Duan.
Molecules 2009 №14, 4351-4357